- Teste de hematologie
- Teste de biochimie
- Biochimie generală din sânge și urina
- Proteine specifice in ser si urina
- Teste biochimice din lichide de punctie
- Teste biochimice din materii fecale
- Teste biochimice pentru tulburari ereditare de metabolism
- Teste pentru nefrolitiaza
- Vitamine, oligoelemente, stres oxidativ
- Acizi grași
- Transferina carbohidrat deficitara (CDT) marker pentru alcoolism
- Markeri non-invazivi pentru afecţiunile hepatice
- Analiza chimică calculi
- Markeri endocrini
- Markeri tumorali
- Markeri virali
- Markeri cardiaci
- Markeri anemie
- Markeri ososi
- Markeri boli autoimune
- Anticorpi antispermatozoizi
- Autoanticorpi in afectiuni endocrine, cardiace, renale
- Autoanticorpi in afectiuni neurologice
- Autoanticorpi in afectiunile dermatologice
- Autoanticorpi in anemia pernicioasa
- Autoanticorpi in diabetul zaharat
- Markeri pentru afectiuni hepatice si gastrointestinale autoimune
- Markeri pentru afectiuni reumatismale si vasculite
- Markeri pentru monitorizarea evolutiei si tratamentului
- Markeri pentru sindromul antifosfolipidic
- Serologie boli infectioase
- Teste specializate de alergologie si imunologie
- Teste de biologie moleculara
- Teste de citogenetica
- Teste de microbiologie
- Toxicologie
- Citologie cervico-vaginala
- Histopatologie
- Consult genetic
- Genetica medicala
- Uncategorized

Boala von Willebrand tip 2N-exoni 17, 24-27
Preț: 1745.00 lei
Informatii generale si recomandari
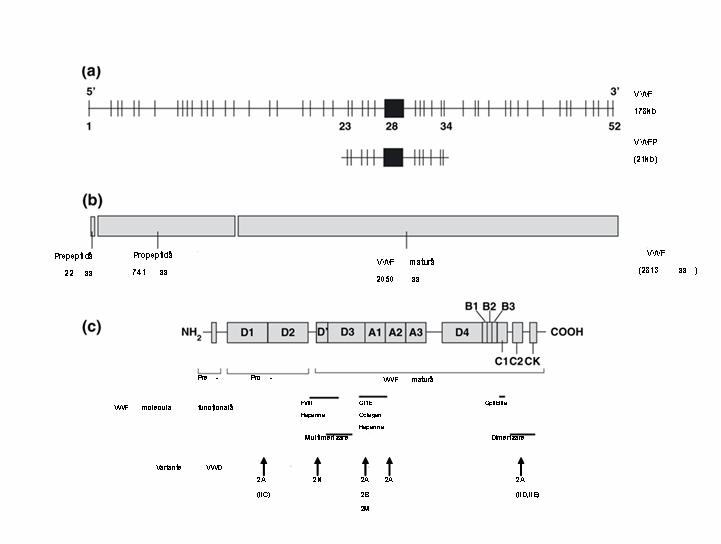
Boala von Willebrand (VWD) este cauzata de deficienta sau anomalia factorului vonWillebrand (FVW) si reprezinta cea mai frecventa boala hemoragica mostenita, cu o prevalenta de 1% in populatia generala. Boala a fost caracterizata pentru prima data de Erick von Willebrand in 1926, ca o conditie patologica mostenita autozomal. VWD se asociaza cu deficiente cantitative (tipul 1 si tipul 3) sau anomalii calitative ale FVW (tipul 2, cu subtipurile 2A, 2B, 2M si 2N). FVW este sintetizat in celulele endoteliale si megakariocite. Gena sa este localizata pe bratul scurt al cromozomului 12p13.3, include ~178 kilobaze cu 52 exoni, cel mai mare exon, 28, avand o lungime de 1.4kb5 si codifica o proteina organizata in 4 domenii repetitive, incluzand 3 “A”, 3 „B”, 2 „C” si 4 „D”, care deservesc functiile diferite ale FVW. Domeniul A1 contine situsurile de legare pentru GPIb plachetara, ristocetina si colagenul tip VI, domeniul A3 contine situsul de legare pentru colagenul I si III, situsul de legare al FVIII este localizat in domeniile D’ si D3, iar domeniul C1 contine secventa RGD capabila de interactiunea cu GPIIb/IIIa plachetara1;2;4.
VWD poate fi mostenita prin mecanisme genetice multiple. Cele mai multe cazuri din tipurile 1 si 2A, precum si tipurile 2B si 2M sunt mostenie autozomal dominant. Tipurile 2N si 3, precum si unele tipuri 1 si 2A sunt mostenite autozomal recesiv. Pentru transmiterea autozomal dominanta, majoritatea indivizilor afectati au un parinte afectat si fiecare copil are 50% sanse sa mosteneasca mutatia. Pentru transmiterea autozomal recesiva parintii sunt heterozigoti obligatorii (carrieri ai unei alele mutante) in general asimptomatici; la conceptie fiecare frate al unui individ afectat are 25% sanse sa fie afectat, 50% sanse sa fie carrier asimptomatic si 25% sanse sa nu fie nici afectat nici carrier, iar fiecare copil al unui individ afectat este heterozigot obligatoriu3.
Mutatiile si polimorfismul genei FvW sunt catalogate intr-o baza de date internationala www.shef.ac.uk/vwf/.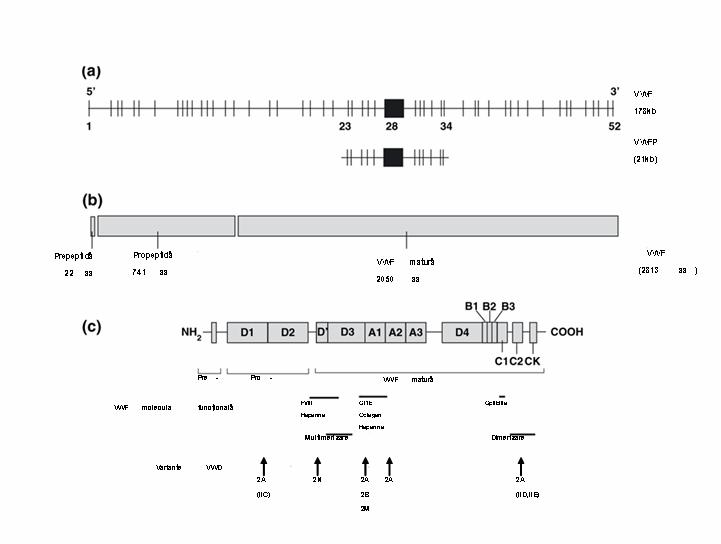
Adaptare dupa Keeney et al5.
Gena factorului vonWillebrand (FVW) domeniile functionale: a) structura genei FVW si a pseudogenei; b) pre-, pro-FVW; c) domeniile functionale si localizarea variantelor tipului 2.
Tipul 1, caracterizat prin deficienta usoara-moderata de FVW, este forma cea mai comuna, fiind responsabil pentru 70% din cazurile de VWD. Majoritatea pacientilor cu tipul 1 de VWD au o boala simptomatica usoara. Nivelul de FVW este scazut, cu reducerea concordanta a FVW:Ag si FVW:RCo, nivelul de FVIII este egal sau mai mare decat cel al FVW si toti multimerii sunt prezenti, daca analiza acestora face parte din evaluarea pacientilor. Criteriile de diagnostic includ un istoric personal si familial de simptome hemoragice, atributabile unor niveluri scazute de FVW <30 UI/dL. Din pacate multi factori fac dificil diagnosticul tipului 1 de VWD. Boala este mostenita ca o tara autozomal dominanta incompleta, cu penetranta variabila, chiar intre membri unei singure familii, astfel ca doar 33% din copii pot fi afectati, iar in unele cazuri de VWD usoara istoricul familial poate sa nu fie pozitiv2;3. Locusuri genetice in afara genei FVW contribuie la variatiile nivelului FVW. Astfel indivizii de grup 0 au niveluri cu 30% mai mici decat cei cu grup sanguin A, B sau AB, existand o prevalenta crescuta printre indivizii de grup 0 de niveluri usor scazute de FVW. De asemenea variatii in nivelurile receptorilor de adeziune plachetari pot modula severitatea simptomelor conferite de deficienta FVW2. Nivelele plasmatice ale FVW pot fi modificate de hormonii tiroidieni, estrogeni, stress8. Recent a fost propus conceptul de “FVW scazut”, ce poate fi aplicat pacientilor cu nivel de FVW sub limita de referinta, dar deasupra unei limite ceva mai mici. Studii ale familiilor pacientilor cu diagnostic de VWD tip 1 sugereaza ca linkajul cu gena FVW este mai putin frecvent intalnit atunci cand nivelul FVW este >30 UI/dL, sugerand ca acest nivel ar putea fi candidatul pentru limita inferioara a categoriei “FvW scazut” 2. Acesti pacienti ar putea avea risc crescut de sangerare fara a fi incadrati ca VWD tip14.
S-au efectuat studii in familii cu tipul 1 de VWD care au avut ca rezultat identificarea prin secventiere a 112 mutatii candidate ca fiind raspunzatoare pentru aparitia sindromului. In plus pacientii cu VWD tip 1 au fost identificati ca fiind purtatori heterozigoti de VWD tip 39. S-au publicat rezultate conform carora o mutatie comuna Tyr1584Cys a fost intalnita la 14% din pacientii canadieni cu VWD tip1 si posibil o proportie similara din pacientii din Marea Britanie4.
Au fost detectate mutatii la 60-65% dintre pacientii cu VWD tipul 1, predominand mutatiile missens; mutatiile missens cu transmitere dominanta complet penetrante sunt adesea identificate cand nivelele FVW:Ag si FVW:RCo sunt <25 UI/dL, in timp ce mutatiile missens mostenite dominant cu penetranta variabila (dominant incompleta), ca de exemplu Tyr1584Cys, sunt identificate la ~50% dintre pacientii cu FVW:Ag si FVW:RCo intre 25-50 UI/dL3.
Mutatiile asociate cu VWD tip 1 afecteaza FVW prin diferite mecanisme incluzand scaderea secretiei si retentia intracelulara datorata alterarii transportului intracelular al subunitatilor FVW, cresterea clearance-ului din circulatie sau, intr-un numar mic de cazuri, reducerea expresiei proteinei rezultata dintr-o alela nula2;3.
Deoarece aproximativ 50% dintre mutatii sunt localizate intre exonii 18-28, initial sunt analizati acesti exoni, secventierea intregii gene furnizand insa informatii complete.
Tipul 3, responsabil pentru 1-5% din cazurile de VWD, este forma cea mai severa, rezultand din deficienta completa a sintezei FVW. Boala hemoragica este in general diagnosticata in copilarie, formarea de hematoame fiind comuna si hemartrozele putand aparea ca urmare a nivelurilor scazute de FVIII intalnite in aceasta forma de boala. La acesti pacienti nivelul FVW este practic nedetectabil, iar nivelul FVIII este in general de 1-10 UI/dL, similar celui din hemofilia A usoara-moderata. Analiza genetica evidentiaza defecte homozigote sau dublu heterozigote ale genei FVW, fiind raportate mutatii la nivelul intregii regiuni codificatoare a genei (exonii 2-52), incluzand deletii mari sau mici ale genei, mutatii ale cadrului de citire, mutatii ale splicing-site (situsului de “innadire”), mutatii missens sau nonsens2;4. Secventierea completa a identificat mutatii in 80-90% din cazurile de VWD tipul 3:
-20% sunt mutatii missens localizate in domeniile D1-D2 (exonii 3-11) si D4-CK (exonii 37-52);
-80% sunt alele nule, localizate la nivelul intregii gene, ce pot rezulta din diferite tipuri de mutatii si care nu produc un produs proteic functional3.
Mutatii ale cadrului de citire si nonsens, care duc la pierderea expresiei proteinei FVW sau la sinteza unei proteine FVW marcat trunchiate, au fost identificate la familii cu VWD tip 3, o mutatie a cadrului de citire la nivelul exonului 18 fiind raspunzatoare de cele mai multe cazuri de VWD tip 3 in populatia suedeza si este comuna in Germania. Aceasta mutatie consta intr-un ARNm stabil care codifica o proteina trunchiata, rapid degradata la nivel celular4.
Deletiile largi ale genei au fost identificate intr-un numar mic de familii, dar acestea confera un risc crescut pentru dezvoltarea de aloanticorpi fata de FVW in urma tratamentului cu concentrate de FVW.
Evaluarea de laborator a parintilor pacientilor cu tipul 3 de VWD poate evidentia o deficienta cantitativa usoara a FVW, dar mai frecvent acestia sunt asimptomatici in concordanta cu un mod autozomal recesiv de transmitere a tipului 3 de VWD. Consanguinitatea este comuna in familiile cu aceasta varianta2;4. Totusi in unele familii cu mutatii nonsens si ale cadrului de citire au fost identificati heterozigoti cu VWD tip 1 aparenta. Mutatii care dau nastere unor subunitati anormale de FVW ce interfera intr-o maniera dominant negativa cu alela normala pot cauza in mod particular VWD simptomatica la heterozigoti4.
Tipul 2 include tipurile calitative de VWD si poate reprezenta pana la 20-25% din cazurile de VWD. Tipul 2 este suspectat cand severitatea simptomelor pacientului apar excesive fata de nivelele de FVW si FVIII, cand exista o reducere discordanta intre FVW:Ag si FVW:RCo sau FVIII sau cand exista concomitent deficienta de FVW si trombocitopenie. In functie de natura defectului functional tipul 2 se clasifica in tipurile 2A, 2B, 2M si 2N2.
–Tipul 2A, intalnit la majoritatea pacientilor cu tipul 2, include formele de VWD caracterizate prin alterarea interactiunii FVW cu trombocitele datorata deficientei formelor cu greutate moleculara mare si intermediara de FVW (care sunt mai potente in interactiunea cu GPIb plachetara si colagenul). Analiza multimerilor FVW evidentiaza reducerea relativa a formelor cu greutate moleculara mare si intermediara. Un fragment proteolitic de 176kD prezent la indivizii normali este marcat crescut la multi pacienti cu VWD tip 2, acest fragment rezultand prin clivajul proteolitic la nivelul situsului Tyr1605-Met1606 de catre ADAMTS13. Este mostenita in general ca o tara autozomal dominanta, generata prin mecanisme genetice multiple. La unii pacienti este alterata multimerizarea FVW (tipul 2A, subgrupul 1), acestia fiind incapabili sa sintetizeze si sa secrete FVW cu lungime completa, mutatiile fiind localizate la nivelul propeptidului FVW (in trecut denumit tipul IIC), regiunii N-terminale a proteinei mature asociate cu formarea multimerilor (tipul IIE) si regiunii C-terminale implicate in formarea initiala a dimerilor (tipul IID). Un al doilea subgrup este caracterizat prin catabolismul rapid de catre ADAMTS13 al multimerilor completi eliberati in plasma. Majoritatea cazurilor de VWD tip 2 sunt mostenite autozomal dominant, fiind raportate rare forme recesive (tipurile IIC si IID). Cele mai multe mutatii sunt grupate in regiunea exonului 28 afectand predominant domeniul A2, incluzand situsul proteolitic Tyr1605-Met1606, si intr-o masura mai mica domeniul A1. Astfel acest exon trebuie examinat primul cand este suspicionat acest subtip de boala. Arg834Trp este mutatia cel mai frecvent intalnita in vWD tipul 2A, absenta multimerilor mari si intermediari fiind efectul proteolizei FVW in plasma. De asemenea mutatii missens au fost raportate in exonii 12-16 (in formele autozomal recesive) si 51-52 (atat in formele autozomal dominante cat si recesive), acestia fiind examinati in continuare2;3;4.
–Tipul 2B este o boala hemoragica paradoxala caracterizata prin interactiunea crescuta a FVW cu trombocitele, urmata de generarea crescuta de complexe FVW-trombocite si clearance-ul acestora din circulatie. Multi din acesti pacienti au trombocitopenie usoara persistenta, care se poate accentua in timpul stresului, sarcinii, interventiilor chirurgicale sau administrarii de desmopresina. Analiza multimerilor evidentiaza, ca si in tipul 2A, absenta formelor cu greutate moleculara mare, iar studiile de agregare plachetara indusa de doze mici de ristocetina evidentiaza interactiunea crescuta a FVW cu plachetele. Tipul 2B este mostenit ca o tara autozomal dominanta, un grup mic de mutatii missens in portiunea exonului 28 care codifica domeniul A1 ce interactioneaza cu GPIb plachetara fiind responsabil pentru toate cazurile raportate2. 4 mutatii grupate intre Arg543 si Arg578 sunt inregistrate la mai mult de 80% din pacienti4. Mutatii care afecteaza p.P1266L pot demonstra doar cresterea legarii GP1b dar fara trombocitopenie si pierderea multimerilor cu greutate moleculara mare3.
–Tipul 2M este caracterizat prin deficienta functiei dependente de plachete a FVW ce nu este atributabila deficientei multimerilor. Ceea ce diferentiaza tipul 2M de tipul 2A este ca analiza multimerilor este normala. Tipul 2M este mostenit ca o tara autozomal dominanta, cu mutatii in regiunea exonului 28 care codifica domeniul A1 al FVW, alterand conformatia proteinei, intr-o arie alternativa fata de tipul 2B2. O mutatie recurenta, Arg1205His – VWD 2M Vicenza a fost recent raportata la familii din Europa, caracterizata prin nivel scazut de FVW:Ag si prezenta de multimeri cu masa moleculara mai mare decat normal (“supranormali”). Boala a fost initial diagnosticata ca VWD tip 1 la pacienti din regiunea Vicenza Italia1;4;8.
–Tipul 2N (anterior denumit tipul Normandy) include mutatii care afecteaza interactiunea FVW cu FVIII, rezultand in „hemofilia autozomala”, cu niveluri de FVIII intre 5 si 30 UI/dL, restul parametrilor de laborator, respectiv FVW:Ag si FVW:RCo, fiind in limite normale. Au fost identificate 37 mutatii4, majoritatea implicand exonii 18, 19 si 20 care codifica o mare parte din domeniul de legare a FVIII si o proportie mult mai mica in exonii 17 si 24-273. Acest tip este mostenit autozomal recesiv, astfel ca pacientii afectati trebuie sa mosteneasca de la parinti doua alele 2N sau o alela 2N de la un parinte si VWD tipul 1 de la celalalt parinte2. Majoritatea indivizilor sunt heterozigoti compusi pentru o mutatie missens si o mutatie care rezulta intr-o alela nula, mai rar sunt heterozigoti compusi pentru 2 mutatii missens, iar o proportie sunt homozigoti pentru o mutatie missens, in mod particular p.Arg854Gln, care apare la 1% din populatia caucaziana3.
Testarea genetica nu este in general necesara pentru stabilirea dignosticului de vWD, dar poate furniza informatii aditionale in ceea ce priveste patogeneza, raspunsul la tratamentul cu desmopresina, consilierea genetica sau riscul formarii aloanticorpilor2.
Testarea genetica este in mod special utila pentru diferentierea unor tipuri 2B de tipul 2A, precum si pentru diagnosticul VWD tipul 2N, respectiv la pacienti cu hemofilie A usoara-moderata cu mostenire atipica sau cu supravietuire scurtata a FVIII infuzat care nu este cauzata de inhibitori specifici sau scaderea activitatii FVIII la femei fara istoric familial de hemofilie A. Initial pacientii trebuie testati pentru mutatii ale genei FVIII, iar cand acestea sunt absente trebuie efectuat screening-ul exonilor FVW3;7.
Testarea pentru carrier a membrilor familiilor cu risc pentru VWD autozomal recesiva este posibila o data ce mutatiile cauzatoare au fost identificate in familie3.
Diagnosticul prenatal pentru sarcinile cu risc crescut (in general pentru tipul 3) este posibil prin analiza ADN-ului extras din celulele fetale obtinute prin amniocenteza in saptamanile 15-18 de gestatie sau din vilozitatile coriale la 10-12 saptamani de sarcina, daca mutatia/mutatiile cauzatoare in familie este/sunt cunoscute sau, in cazul in care mutatiile nu sunt cunoscute, diagnosticul poate fi incercat prin analiza linkajului genetic utilizand un panel larg al polimorfismelor cunoscute in gena FVW, daca structura familiala este suficienta, rezultatele putand fi obtinute mai rapid decat identificarea mutatiilor in ambele alele3;4.
Specimen recoltat – sange venos6.
Recipient de recoltare – vacutainer ce contine EDTA ca anticoagulant6.
Cantitate recoltata – cat permite vacuumul6.
Cauze de respingere a probei – folosirea heparinei ca anticoagulant6.
Stabilitate proba – 7 zile la 2-8ºC6.
Metoda
Sunt disponibile urmatoarele variante de testare:
-VWD tip 2N – secventierea exonilor 17, 24-27 (pot fi identificate restul mutatiilor) 6.
Limite si interferente
Clasificarea actuala nu restrictioneaza VWD ca fiind cauzata de mutatii localizate la nivelul genei FVW. Evaluarea intregii gene nu poate identifica existenta unei mutatii in cazuri de VWD aparenta. Astfel imposibilitatea identificarii unei mutatii cauzatoare nu exclude diagnosticul de VWD.
Variantele normale ale genei FVW sunt foarte comune, actual fiind cunoscute aproximativ 150 variante normale. Acest grad crescut al polimorfismului, impreuna cu dimensiunile mari ale genei si prezenta unei pseudogene partiale (localizata pe cromozomul 22q si corespunzand exonilor 23-34) pot face dificila secventarea completa a genei si interpretarea datelor3;8. In plus, defectele genetice din deficientele cantitative ale FVW sunt distribuite in intreaga gena, complicand evaluarea.
Tipul plachetar de VWD rezulta din mutatii in GP1BA, dar se prezinta fenotipic ca VWD tipul 2B.
Sindromul vonWillebrand dobandit este o boala hemoragica usoara-moderata care nu este cauzata de o mutatie a genei FVW, cel mai adesea intalnita la persoane peste 40 ani fara istoric de sangerare si care este asociata cu o varietate de conditii: sindroame limfoproliferative, paraproteinemii, boli autoimune, sindrom antifosfolipidic, proteoliza crescuta a FVW datorata unor modificari conformationale induse de forte de frecare crescute (stenoza aortica, defect septal ventricular), trombocitoza marcata, indepartarea FVW din circulatie datorita legarii de celule tumorale (tumora Wilm`s sau anumite boli limfoproliferative), scaderea sintezei FVW (hipotiroidism), medicamente (acid valproic, ciprofloxacin, griseofulvin)3.
Bibliografie
1. Castaman G, Tosseto A, Rodeghiero F. „von Willebrand Disease”. In Practical Hemostasis and Thrombosis, O’Shaughnessy D, Makris M, Lillicrap D eds, Blackwell Publishing, 2008, 53-54.
2. Friedman K, Rodgers G. „Inherited Coagulation Disorders”. In Wintrobe`s Clinical Hematology, Greer J, Foerster J, Lukens J, Rodgers G, Pareskevas F, Glader B, 11th ed, Lippincott Williams & Wilkins, 2004, 1392-94.
3. Goodeve A, James P. „von Willebrand Disease”. GeneReviews, NCBI Bookshelf.
4. Johnsen J, Ginsburg D. „von Willebrand Disease”. In Williams Hematology, Lichtman M, Beutler E, Kipps T, Seligsohn U, Kaushansky K, Prchal J., 7th ed, McGraw-Hill Medical, 2006, 1929-37.
5. Keeney S, Bowen D, Cumming A, Enayat S, Goodeve A, Hill M. „The molecular analysis of von Willebrand disease: a guideline from the UK Haemophilia Centre Doctors’ Organisation Haemophilia Genetics Laboratory Network”. Haemophilia, (2008); 14: 1099–111.
6. Laborator Synevo. Referintele specifice tehnologiei de lucru utilizate, 2010. Ref Type: Catalog.
7. Mayo Clinic, Mayo Medical Laboratories. Reference Laboratory Services for Health Care Organizations. von Willebrand Disease 2N (subtype Normandy), Blood. www.mayomedicallaboratories.com. 2010. Ref Type: Internet Communication.
8. Online Mendelian Inheritance in Man, McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine. „von Willebrand Disease”.
9. Rajiv K. Pruthi, MBBS, “A Practical Approach to Genetic Testing for vonWillebrand Disease”, Mayo Clinic Proceedings (May 2006) 81;5: 679-691.
Produsul a fost adăugat în coș
În plus, ai la dispoziție 30 de zile pentru a veni la recoltare.