înapoi la lista
Hemofilia B-testare genetica
Inchide
Informatii generale Hemofilia B-testare genetica
Hemofilia B (boala Christmas), este caracterizata prin deficienta activitatii coagulante a FIX si este mostenita ca o tara recesiva X-linkata, gena FIX (F9) fiind situata centromeric fata de gena FVIII pe cromozomul X, la Xq27. Prevalenta hemofiliei B este de 1/20-30 mii nou-nascuti vii de sex masculin, hemofilia A fiind de 4-8 ori mai frecventa. Ca si hemofilia A, hemofilia B este prezenta la toate grupurile etnice1;2;5.
Boala se manifesta la barbati care nu prezinta o alela normala; acestia nu transmit anomalia la fiii lor, dar toate fiicele sunt purtatoare ale tarei. Majoritatea femeilor carrier nu sunt afectate datorita prezentei unei alele normale mostenite de la mama, iar ele vor transmite boala la jumatate din fiii lor si statusul de carrier la jumatate din fiice.
Fenotipic boala este similara hemofiliei A, defectele FIX putand fi severe sau usoare, formele severe fiind mai rare dacat in hemofilia A. Pacientii cu hemofilie B severa (nivel de FIX <1%) sunt diagnosticati in primul an de viata, sangerarile musculare profunde si hemartrozele spontane reprezentand simptomele cele mai frecvente. Indivizii cu hemofilie B moderat severa (FIX = 1-5%) prezinta de obicei sangerari prelungite sau intarziate dupa traume minore si sunt diagnosticati in general inainte de varsta de 5-6 ani. Indivizii cu hemofilie B usoara (FIX = 5-30%) prezinta sangerari anormale dupa interventii chirurgicale, extractii dentare sau traume majore, acestia fiind de obicei diagnosticati mai tarziu in timpul vietii1.
La orice individ cu hemofilie B frecventa episoadelor hemoragice poate fi mai mare in copilarie si adolescenta decat la varsta adulta. Aproximativ 10% din femeile carrier prezinta risc de sangerare, avand nivele de FIX <30%, indiferent de severitatea hemofiliei B in familie, simptomele fiind de obicei usoare. O crestere foarte usoara a simptomelor hemoragice poate exista chiar la cele cu activitate coagulanta a FIX de 40-60%. Sangerarea poate fi mai severa la cele cu activitatea FVIII la limita inferioara a normalului1. Variatiile in nivelul FIX sunt atribuite lionizarii (inactivarea cromozomului X normal in cursul embriogenezei)1;6.
Alte cauze ale hemofiliei A la femei sunt: copilul de sex feminin mosteneste doi cromozomi X anormali de la tatal hemofilic si mama carrier; anumite boli cromozomiale pot duce la aparitia unui genotip hemizigot si induc hemofilia prin absenta unei alele normale: mozaicism 45XX/45X, cariotip 46XY, deletia cromozomului X (genotip XO, Sindrom Turner). In cazuri rare bolnavi nehemofilici, cu boli autoimune (lupus eritematos sistemic, artrita reumatoida) sau dupa administrare de medicamente (peniciline, fenitoin) sau cu boli limfoproliferative, pot dezvolta autoanticorpi anti-FIX care determina aparitia hemofiliei B dobandite.
In functie de detectia antigenului FIX in plasma, pot fi definite 3 grupuri distincte: o varianta CRM (cross reactive material) – pozitiva, forma cea mai comuna, o varianta CRM-negativa si o varianta CRM-redusa, in care nivelul antigenului si activitatii coagulante a FIX sunt proportional reduse2;6.
Modificarile de laborator clasice sunt reprezentate de aPTT prelungit cu PT normal. Un subset de pacienti cu hemofilie B prezinta PT prelungit atunci cand se utilizeaza creier de origine bovina ca sursa de tromboplastina, acesti pacienti CRM (+) fiind clasificati ca avand hemofilie B(M). Un numar de mutatii missense care afecteaza aminoacizii din pozitiile 180, 181, 182 si cateva reziduuri din apropierea situsului activ rezulta intr-un FIX anormal si inactiv care actioneaza ca inhibitor al reactiei normale dintre FVII si FX si creierul bovin. PT determinat cu creier de iepure sau tromboplastina de origine umana nu este prelungit2;6.
In hemofilia B Leyden manifestarile clinice tind sa diminueze o data cu inaintarea in varsta in asociere cu cresterea nivelului FIX de la ~1UI/dL in copilarie pana la 20UI/dL in viata adulta. Varianta Leyden este caracterizata ca CRM (-) la nastere, devenind ulterior CRM (+) sau redusa. Baza genetica a acestei variante este faptul ca mutatiile responsabile sunt localizate la nivelul promotorului genei FIX, la bp -23 pana la bp +13, care se crede ca introduc in promotor un element responsiv la androgeni si care, cu varsta, stimuleaza transcriptia si sinteza proteinei2;4.
Rogaev et al.(2009) au identificat o mutatie a situsului de splicing la nivelul genei F9 ca mutatia cauzativa pentru ‘boala regala’, forma de hemofilie transmisa de la Regina Victoria la familiile regale europene si transmisa nepoatei sale, Imparateasa Rusiei Alexandra, si fiului acesteia, printul Alexei6.
Gena FIX este considerabil mai mica decat gena FVIII, avand 34 kb lungime, continand numai 8 exoni care codifica un ARNm de 2.8 kb. Ca si FVIII, FIX este sintetizat predominant in ficat si este omolog FVII, FX si PC. Produsul genei include cateva domenii distincte: primul si al doilea domeniu reprezinta peptidul semnal si, respectiv, propeptidul, care sunt clivate generand proteina matura de 415 aminoacizi.
Modificarile posttranslationale includ glicozilare, sulfatare, fosforilare, β-hidroxilare si γ-carboxilare. O γ-carboxilaza se leaga la propeptid inaintea clivarii si, intr-o etapa dependenta de vitamina K, converteste primele 12 reziduuri de acid glutamic de la capatul N-terminal in reziduuri γ-carboxilglutamice sau Gla. Apoi domeniul Gla leaga ionii de Ca si adopta o conformatie capabila de legarea la suprafetele fosfolipidice unde are loc coagularea. Adiacent domeniului Gla sunt doua domenii omologe factorului de crestere epidermal. Urmatoarele domenii constituie o secventa de conectare care include peptidul activator, iar in final domeniul catalitic, acesta fiind o serinproteaza tipica. Peptidul activator este clivat de la nivelul formei de zimogen a FIX de catre FVIIa/FT sau FXIa, rezultand enzima activa FIXaβ1;4;7.
Rar, mutatii la nivelul domeniului de legare a carboxilazei determina cresterea sensibilitatii la tratamentul cu warfarina la indivizi fara tendinta hemoragica de baza1.
Au fost descrise 10 polimorfisme utile pentru analiza linkajului genetic asociate cu gena F9. O lista actualizata a mutatiilor este disponibila pe www.kcl.ac.uk/ip/petergreen/haemB-database.html.
Diagnosticul de hemofilie B este stabilit la indivizi cu nivel scazut al activitatii coagulante a FIX1.
Deficienta usoara a FIX trebuie intotdeauna luata in considerare in diagnosticul diferential al pacientilor cu sangerari sugestive si rezultate normale ale testelor de coagulare de rutina (PT, aPTT), deoarece multi reactivi pentru aPTT nu detecteaza deficientele usoare (20-30%) ale FIX1;2.
Activitatea coagulanta a FIX in sangele de cordon la nou-nascutii la termen este mai mica decat la adulti (in medie ~30%, limite 15-50%); astfel diagnosticul de hemofilie B poate fi stabilit la sugar daca activitatea FIX <1%, dar este echivoc la valori moderat scazute (15-20%), iar testul trebuie repetat la varsta de 6 luni1;4.
Recomandari pentru testarea genetica in hemofilia A
- Testarea genetica a unui pacient pentru detectia mutatiei specifice a FIX intr-o familie, in vederea obtinerii de informatii necesare consilierii genetice a membrilor familiei cu risc crescut.
- La indivizii care reprezinta singurul caz din familie identificarea mutatiei specifice poate ajuta la predictia fenotipului clinic si stabilirea riscului de a dezvolta inhibitori ai FIX.
- Testarea pentru carrier a rudelor cu risc (necesita identificarea mutatiei cauzatoare in familie) este recomandata inaintea sarcinii sau cat mai devreme in sarcina.
- Diagnosticul prenatal si preimplantare pentru sarcinile cu risc (necesita identificarea mutatiei cauzatoare in familie) Procedura uzuala consta in determinarea sexului fetal prin analiza cromozomilor in celulele fetale obtinute din vilozitatile coriale la 10-12 saptamani de sarcina sau prin amniocenteza la 15-18 saptamani, iar daca carioptipul este 46XY, ADN-ul extras din celulele fetale poate fi analizat pentru prezenta mutatiei sau pentru markerii informativi Daca mutatia nu este cunoscuta, iar linkajul nu este informativ, diagnosticul prenatal este posibil prin masurarea activitatii coagulante a FIX in sangele obtinut prin punctia percutana a cordonului ombilical la 18-21 saptamani de sarcina (diagnosticul de hemofilie B poate fi stabilit daca nivelul FIX este <1%, dar este echivoc daca este moderat scazut)1.
– sange venos3.
Recipient de recoltare – vacutainer ce contine EDTA ca anticoagulant3.
Cantitate recoltata – cat permite vacuumul3.
Cauze de respingere a probei – folosirea heparinei ca anticoagulant3.
Stabilitate proba – 7 zile la 2-8ºC3.
Metoda – secventierea genei F9 si analiza deletiilor/duplicatiilor prin MLPA3.
Interpretarea rezultatelor
Aproximativ 99% din pacientii cu hemofilie B prezentand mutatii la nivelul genei F9. Au fost descrise 896 mutatii distincte raportate in baza de date a FIX, ~30% aparand la nivelul dinucleotidelor CpG, care de obicei implica reziduuri de arginina critice, rezultand in proteine disfunctionale. Diferite genotipuri se asociaza cu lipsa absoluta sau relativa a proteinei FIX, iar cateva mutatii missense se asociaza cu o proteina disfunctionala7. Cel mai frecvent intalnite sunt mutatiile missense (54%) urmate de mutatiile STOP (21,3%).
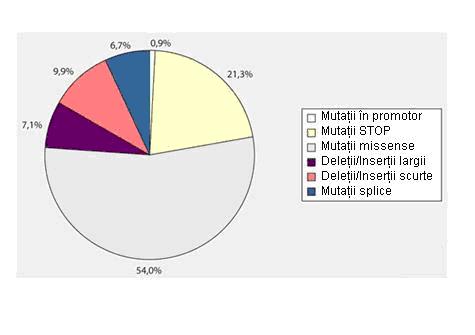

Adaptare dupa Oldenburg et al., Medizinische Genetik, 2008
Distributia procentuala a mutatiilor la pacientii cu hemofilie B.
Hemofilia B severa este cauzata de alterari severe ale genei, incluzand mutatii ale cadrului de citire, ale situsului de splicing, mutatii nonsens sau missens. Ocazional indivizii cu hemofilie B severa prezinta deletii exonice, multiexonice sau deletii complete ale genei. Hemofilia B usoara-moderata se asociaza de regula cu mutatii missens.
Mutatiile punctiforme sunt responsabile de majoritatea mutatiilor de novo. Deletiile largi partiale sau complete sau mutatii nonsens care determina lipsa FIX antigen circulant, cum ar fi p.Arg29X (c.85C>T), se asociaza cu aparitia inhibitorilor (1-3% din pacientii cu hemofilie B severa)1;5.
Inhibitorii fata de FIX apar numai la indivizii cu boala Christmas care nu au FIX antigen detectabil, dar spre deosebire de FVIII inhibitorii fata de FIX apar mai putin frecvent, sugerand ca poate exista o predispozitie pentru aparitia acestora6. Recent s-a observat ca o parte din acesti pacienti prezinta risc de reactii anafilactice la administrarea de tratament substitutiv cu FIX5.
Exista elemente ale istoricului familial predictoare pentru statusul de carrier al unei paciente:
- o femeie care are un fiu afectat si o alta ruda afectata pe linie materna este carrier obligatoriu;
- daca o femeie are mai mult de un fiu afectat si mutatia nu poate fi detectata la nivelul ADN-ului sau, atunci ea prezinta mozaicism al liniei germinale.
Aproximativ o treime din mutatiile responsabile de hemofilia B sunt mutatii de novo7. Daca un pacient este un caz simplex (fara istoric familial de hemofilie), exista cateva posibilitati in ceea ce priveste statusul de carrier al mamei sale:
- mama nu este carrier si pacientul prezinta o mutatie de novo;
- mama este carrier de novo mutatia aparand ca o mutatie a liniei germinale (prezenta in toate celulele si detectabila la nivelul ADN-ului), ca o mutatie somatica (prezenta in unele dar nu in toate celulele si care poate fi detectata in ADN-ul leucocitar in ≤11% din familii) sau mozaicism al liniei germinale (mutatia este prezenta in unele celule germinale, dar nu este detectabila in ADN-ul din leucocite);
- mama este carrier si a mostenit mutatia cauzatoare de la mama care are o mutatie de novo sau de la tatal asimptomatic care este mozaic pentru mutatie;
- mama este carrier pentru o mutatie aparuta in generatiile anterioare, care a trecut prin familie ,fiind asimptomatica la femeile carrier.
Testarea ADN combinata cu analiza linkajului poate adesea determina punctul de origine al unei mutatii de novo, importanta pentru determinarea ramurilor din familie cu risc de hemofilie B1.
Mozaicismul liniei germinale este posibil, dar rar; astfel daca un individ afectat se prezinta ca un caz simplex, iar mutatia fiului nu poate fi detectata in ADN-ul din leucocitele mamei, aceasta prezinta totusi un risc (desi mic) de a avea si alti copii afectati1.
Limite si interferente
Cand testarea pentru carrier este efectuata fara identificarea anterioara a mutatiei in familie, obtinerea unui rezultat negativ la o ruda cu risc nu exclude un potential carrier.
Activitatea coagulanta a FIX este normala la majoritatea carrier-lor si astfel nu poate fi utilizata pentru determinarea statusului de carrier, testarea genetica fiind esentiala in acest caz1;5.
Bibliografie
1. Brower C, Thompson A. „Hemophilia B”. GeneReviews, NCBI Bookshelf. Ref Type: Internet Communication.2. Friedman K, Rodgers G. „Inherited Coagulation Disorders”. In Wintrobe`s Clinical Hematology, Greer J, Foerster J, Lukens J, Rodgers G, Pareskevas F, Glader B, 11th ed, Lippincott Williams & Wilkins, 2004, 1395-96.3. Laborator Synevo. Referintele specifice tehnologiei de lucru utilizate, 2010. Ref Type: Catalog.4. Maclean R, Makris M. „Hemophilia A and B”. In Practical Hemostasis and Thrombosis, O’Shaughnessy D, Makris M, Lillicrap D eds, Blackwell Publishing, 2008, 42-43.5. Mayo Clinic, Mayo Medical Laboratories. Reference Laboratory Services for Health Care Organizations. “Hemophilia B, FactorIX Gene Mutation Screening”. www.mayomedicallaboratories.com. 2010. Ref Type: Internet Communication.6. Online Mendelian Inheritance in Man, McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine. „Hemophilia B”. Ref Type: Internet Communication.7. Roberts H, Escobar M, White II G. In Williams Hematology, Lichtman M, Beutler E, Kipps T, Seligsohn U, Kaushansky K, Prchal J., 7th ed, McGraw-Hill Medical, 2006, 1880-1881.Vezi tot conținutul
Vezi mai puțin
