- Teste de hematologie
- Teste de biochimie
- Biochimie generală din sânge și urina
- Proteine specifice in ser si urina
- Teste biochimice din lichide de punctie
- Teste biochimice din materii fecale
- Teste biochimice pentru tulburari ereditare de metabolism
- Teste pentru nefrolitiaza
- Vitamine, oligoelemente, stres oxidativ
- Acizi grași
- Transferina carbohidrat deficitara (CDT) marker pentru alcoolism
- Markeri non-invazivi pentru afecţiunile hepatice
- Analiza chimică calculi
- Markeri endocrini
- Markeri tumorali
- Markeri virali
- Markeri cardiaci
- Markeri anemie
- Markeri ososi
- Markeri boli autoimune
- Anticorpi antispermatozoizi
- Autoanticorpi in afectiuni endocrine, cardiace, renale
- Autoanticorpi in afectiuni neurologice
- Autoanticorpi in afectiunile dermatologice
- Autoanticorpi in anemia pernicioasa
- Autoanticorpi in diabetul zaharat
- Markeri pentru afectiuni hepatice si gastrointestinale autoimune
- Markeri pentru afectiuni reumatismale si vasculite
- Markeri pentru monitorizarea evolutiei si tratamentului
- Markeri pentru sindromul antifosfolipidic
- Serologie boli infectioase
- Teste specializate de alergologie si imunologie
- Teste de biologie moleculara
- Teste de citogenetica
- Teste de microbiologie
- Toxicologie
- Citologie cervico-vaginala
- Histopatologie
- Consult genetic
- Genetica medicala

Imunofenotipare limfocitara – sindrom limfoproliferativ
Preț: 1036.00 lei
Această analiză se poate recolta în centrele Synevo, conform unui program special:
– In centrele de recoltare din București/ Ilfov, Pitești, Târgoviște, Râmnicu Vâlcea, Alexandria, Giurgiu Buzău și Urziceni de luni până joi;
– In centrele de recoltare din restul țării, in zilele de luni si miercuri.
Informaţii generale Imunofenotipare limfocitara – sindrom limfoproliferativ
Imunofenotiparea prin citometrie in flux (IFCF) reprezinta un instrument indispensabil pentru diagnosticul, clasificarea, stadializarea si monitorizarea neoplaziilor hematologice.
La Conferinţa Internaţionala de Consens din 2006 de la Bethesda, Maryland s-au emis recomandari privind indicaţiile medicale ale testarii prin citometrie in flux. Astfel, aceasta este indicata in urmatoarele situaţii clinice:
- citopenii, in special bicitopenia si pancitopenia;
- numar crescut de leucocite, incluzand limfocitoza, monocitoza si eozinofilia;
- prezenţa de celule atipice sau blasti in sangele periferic, maduva osoasa sau alte lichide ale corpului;
- plasmocitoza sau gamopatia monoclonala;
- organomegalia si masele tisulare.
In aceste situaţii imunofenotiparea poate constitui un screening sensibil pentru prezenţa/absenţa unei neoplazii hematologice. In plus, citometria in flux este un instrument util in stadializarea unei neoplazii limfoide diagnosticate anterior, in monitorizarea raspunsului la tratament incluzand detectarea bolii minime reziduale, in documentarea recaderii sau progresiei si in diagnosticul unei malignitaţi hematologice intercurente.
In malignitaţile hematologice mai mulţi pasi sunt urmaţi in aplicarea si interpretarea informaţiei imunofenotipice:
- identificarea celulelor din linii diferite si determinarea daca acestea sunt mature sau imature;
- detectarea celulelor anormale prin identificarea expresiei antigenice care difera semnificativ de normal;
- documentarea detaliata a fenotipului populaţiei celulare anormale (adica prezenţa sau absenţa antigenelor) si, prin comparaţie cu populaţia normala, documentarea unei cresteri sau scaderi a intensitaţii colorarii cu anticorpi marcaţi cu fluorocromi;
- evaluarea faptului daca informaţiile obţinute sunt diagnostice pentru o anumita boala, iar daca nu, stabilirea unei liste de entitaţi posibile cu sugerarea unor studii suplimentare ce pot avea valoare diagnostica, cum ar fi imunohistochimie (IHC), citogenetica, FISH sau studii moleculare;
- furnizarea de informaţii imunofenotipice ce pot avea valoare prognostica suplimentara, incluzand identificarea de ţinte pentru o potenţiala terapie ţintita.
Atunci cand se primeste o proba pentru testare prin citometrie in flux, se stabilesc liniile celulare si antigenele ce trebuie evaluate pe baza tipului de proba si alte informaţii disponibile, cum ar fi: indicaţia medicala pentru testare, istoricul clinic, elementele morfologice, testarile anterioare prin citometrie in flux, rezultatele altor teste de laborator, precum si rezultatele unei eventuale testari preliminare sceening prin citometrie in flux. Pentru indicaţiile medicale identificate de grupul Bethesda s-a stabilit un consens privind liniile celulare care trebuie evaluate si antigenele incluse in evaluarea primara a fiecarei linii1.
Astfel reactivii de consens menţionaţi in tabelul 17.9.5.1, atunci cand sunt evaluaţi in combinaţiile potrivite, reprezinta minimul care permite evaluarea eficienta a liniilor celulare indicate pentru neoplazia hematopoietica cu un grad acceptabil de sensibilitate2.
Tabel 17.9.5.1: Reactivii consens pentru evaluarea iniţiala a neoplaziilor hematopoietice16
|
Linia celulara |
Reactivii primari |
|
Celulele B |
CD5, CD10, CD19, CD20, CD45, Kappa, Lambda |
|
Celulele T si NK |
CD2, CD3, CD4, CD5, CD7, CD8, CD45, CD56 |
|
Celulele mielomonocitare |
CD7, CD11b, CD13, CD14, CD15, CD16, CD33, CD34, CD45, CD56, CD117, HLA-DR |
|
Celulele mielomonocitare (panel limitat) |
CD13, CD33, CD34, CD45 |
|
Plasmocite |
CD19, CD38, CD45, CD56 |
Este furnizat de asemenea un set secundar de reactivi potrivit pentru caracterizarea in situaţii specifice, dar numai un numar limitat dintre acestia va fi utilizat intr-un caz particular (vezi tabelul 17.9.5.2). De menţionat faptul ca in condiţii practice specifice poate fi necesara inlocuirea anumitor reactivi intre cele doua paneluri pentru adaptarea la o anumita populaţie de pacienţi sau o anumita prevalenţa a bolilor, dar numarul total al reactivilor utilizaţi trebuie sa ramana acelasi.
Tabel 17.9.5.2: Reactivi pentru evaluarea secundara a liniilor celulare hematopoietice specifice16
| Linia celulara |
Reactivii secundari
|
| Celulele B | CD9, CD11c, CD15, CD22, cCD22, CD23, CD25, CD13, CD33, CD34, CD38, CD43, CD58, cCD79a, CD79b, CD103, FMC7, Bcl-2, cKappa, cLambda, TdT, ZAP-70, cIgM |
|
Celulele T si NK |
CD1a, cCD3, CD10, CD16, CD25, CD26, CD30, CD34, CD45RA, CD45RO, CD57, αβ-TCR, γδ-TCR, cTIA-1, T – izoformele lantului beta, TdT |
|
Celulele mielomonocitare |
CD2, CD4, CD25, CD36, CD38, CD41, CD61, cCD61, CD64, CD71, cMPO, CD123, CD163, CD235a |
|
Plasmocite |
CD10, CD117, CD138, cKappa, cLambda |
In prezent imunofenotiparea limfocitelor din sangele periferic este utilizata in mod comun in evaluarea pacienţilor cu limfocitoza susţinuta pentru determinarea naturii sale reactive sau maligne3.
NEOPLAZIILE LIMFOIDE MATURE
Acestea includ leucemia limfocitara cronica si limfoamele non-Hodgkin, recunoscute dupa un fenotip care este similar celulelor limfoide mature normale (cum ar fi prezenţa Ig de suprafaţa pe celulele B mature) si lipsa trasaturilor antigenice de imaturitate (cum ar fi expresia TdT, CD34 si intensitate scazuta a expresiei CD45). Prin identificarea antigenelor asociate liniei celulare, neoplaziile limfoide mature pot fi imparţite in neoplazii ale liniei celulare B, T sau NK.
Neoplaziile limfoide cu celula B matura
Citometria in flux este indispensabila pentru diagnosticul neoplaziilor limfoide cu celula B matura, prin identificarea celulelor anormale fenotipic aparţinand liniei celulare B si recunoasterea fenotipurilor caracteristice diferitelor entitaţi2. Multe cazuri de posibil limfom cu celula B sunt evaluate iniţial prin imunofenotiparea sangelui periferic si/sau aspiratului de maduva osoasa inaintea efectuarii biopsiei tisulare. Limfoamele cu celula B identificate astfel sunt adesea incadrate in spectrul larg al bolilor limfoproliferative cronice cu celula B (BLPC-B), aceasta terminologie fiind utilizata deoarece subclasificarea precisa utilizand criteriile OMS necesita corelarea cu histopatologia tisulara. In ciuda acestei cerinţe, necesitatea unui diagnostic specific rapid, utilizand proceduri minim invazive, a dus la cresterea tendinţei de subclasificare completa a BLPC-B care afecteaza sangele periferic si maduva osoasa in mare parte pe rezultatele IFCF sau in combinaţie cu histopatologia medulara. Aceasta tendinţa a fost impulsionata de studii ce au documentat identitatea fenotipica in neoplaziile cu celula B cu afectare concomitenta a sangelui periferic sau maduvei osoase si ganglionilor limfatici13.
In plus citometria in flux poate fi utilizata pentru identificarea expresiei unor ţinte pentru terapia cu anticorpi si furnizarea de informaţii prognostice suplimentare. De asemenea, in urma terapiei, citometria de flux constituie metoda stabilita pentru evaluarea bolii minime reziduale2.
Celulele limboide B mature neoplazice pot fi distinse de celulele normale prin identificarea a 2 tipuri principale de anomalii fenotipice: restricţia clasei lanţurilor usoare de Ig si expresia aberanta de antigene2;11.
Spre deosebire de majoritatea populaţiilor normale sau reactive, neoplaziile cu celula B matura reprezinta de obicei o singura clona celulara care exprima o singura clasa de lanţ usor de Ig, adica kappa sau lambda, reflectata printr-un raport anormal kappa-lambda. In mod tipic, leucemia limfocitara cronica (CLL) prezinta o intensitate scazuta a colorarii pentru Ig de suprafaţa. Unele celule B nu prezinta Ig de suprafaţa, acestea incluzand limfoblastii, plasmocitele si celulele B timice, precum si corespondenţele lor neoplazice: leucemia acuta limfoblastica, neoplaziile plasmocitare si limfomul cu celula B mediastinal primar. Rezultate fals negative pentru Ig de suprafaţa se pot datora interferenţei Ac solubili cu legarea Ac de detecţie sau alterarii/deleţiei epitopilor recunoscuţi, datorate de exemplu mutaţiilor hipersomatice din limfomul folicular.
Prin IFCF se poate identifica expresia aberanta de antigene la nivelul celulelor B. Expresia CD5 la nivelul celulelor B este in general raportata ca un fenotip aberant, dar exista populaţii mici de celule B normale mature CD5+, aflate de obicei in sangele periferic, dar si la nivelul ganglionilor limfatici, in special la pacienţii cu boli autoimune. Expresia CD5 a fost raportata si la nivelul unor precursori normali ai celulelor B din maduva osoasa. Astfel expresia CD5 trebuie interpretata in corelaţie cu alte anomalii, incluzand restricţia lanţului usor de Ig sau alterarea intensitaţii colorarii CD20, CD22 si CD79b. Un alt tip de aberaţie fenotipica este expresia de antigene care nu sunt in mod tipic prezente intr-un subset de celule B aparţinand unui compartiment biologic distinct (de exemplu expresia bcl-2 pe celulele B CD10+)2.
Intr-un studiu, Morice si colaboratorii au evaluat retrospectiv abilitatea IFCF a sangelui periferic si maduvei osoase de a prezice tipul de limfom cu celula B intr-un grup de 252 pacienţi cu IFCF pozitive si biopsii tisulare diagnostice, iar concluziile extrase din acest studiu corelativ sunt rezumate intr-un algoritm pentru clasificarea limfoamelor cu celula B prin IFCF a sangelui periferic/maduvei osoase (vezi figura 17.9.5.1)13.
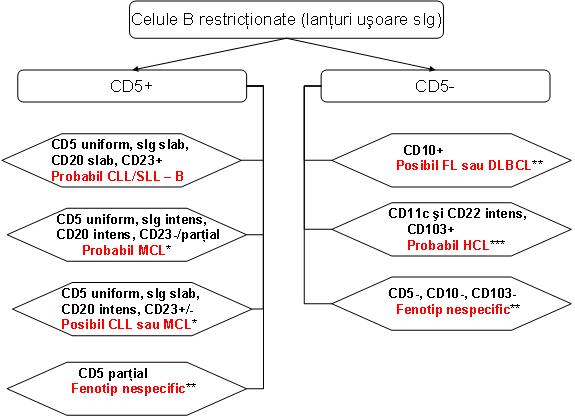
Fig. 17.9.5.1 Adaptare dupa Morice W G et al. Mayo Clin Proc. 2008; 83:776-785
Imunofenotiparea prin citometrie in flux este in mod particular utila pentru subtiparea limfoamelor/leucemiilor cu celula B compuse predominant din celule mici (vezi tabelul 17.9.5.3)5.
Tabel 17.9.5.3
|
Antigenele de suprafaţa in limfoamele/leucemiile cu celula B mica |
|||||||
|
NHL |
sIg |
CD5 |
CD10 |
CD23 |
CD11c |
CD103 |
CD25 |
|
CLL/SLL |
Slab |
+ |
– |
++/+ |
– |
– |
– |
|
MCL |
+ |
+ |
-/+ |
-/+ |
– |
– |
– |
|
Folicular |
+ |
– |
+ |
+/- |
– |
– |
– |
|
LPL |
+ |
– |
– |
– |
– |
– |
– |
|
MZL |
+ |
– |
– |
– |
+/- slab |
– |
– |
|
SMZL |
+ |
– |
– |
– |
+/- slab |
+/- slab |
– |
|
Hairy cell |
+ |
– |
-/+ |
– |
++ |
+ |
+ |
NHL = limfom non-Hodgkin, sIg = imunoglobuline de suprafata, CLL = leucemie limfocitara cronica, SLL = limfom cu limfocite mici, LPL = limfom limfoplasmocitar, MCL = limfom/leucemie cu celula a mantalei, MZL = limfom de zona marginala, SMZL = limfom de zona marginala splenic
▪ Expresia aberanta a CD5 este caracteristica pentru CLL/SLL si MCL, care prezinta manifestari superpozabile. Coexpresia omogena a CD5 si CD23 de catre o BLPC-B, atunci cand este insoţita de colorarea slaba atat pentru sIg cat si pentru CD20, CD22, CD79b, iar FMC-7– prezice CLL/SLL cu specificitate inalta. Acest fenotip exclude MCL (in care de obicei CD23 este negativ), iar alte limfoame similare fenotipic sunt relativ indolente, astfel incat efectele clinice ale clasificarii unuia din aceste cazuri in categoria CLL ar fi probabil minime. Totusi acest diagnostic IFCF trebuie confirmat prin corelarea cu date clinice, de laborator si citologice, cu recomandarea de biopsie tisulara cand subclasificarea precisa ramane neclara13.
Un fenotip caracterizat prin expresia intensa a sIg si CD20, pozitivitate CD5 uniforma, iar expresia CD23 fie absenta fie parţiala si FMC-7+ se asociaza puternic cu MCL2;13.
In anumite cazuri IFCF prezinta aspecte care se suprapun intre fenotipurile CLL si MCL, expresia slab pozitiva a CD23 putand fi intalnita in ambele. Astfel, dat fiind comportamentul clinic relativ agresiv al MCL, studii suplimentare, care includ imunohistochimie pentru supraexpresia ciclinei D1 sau examen citogenetic pentru identificarea translocaţiei t(11;14)(q13;q32) sau FISH pentru gena de fuziune CCND1/IGH, sunt necesare pentru diagnostic in toate cazurile de BLPC-B care prezinta expresia uniforma a CD5 si in care lipsesc alte atribute fenotipice tipice pentru CLL. Totusi absenţa genei de fuziune in aceste cazuri nu stabileste diagnosticul definitiv de CLL, alte boli (LPL, SMZL) putand prezenta un imunofenotip similar. De asemenea in cazurile cu IFCF tipica pentru MCL un FISH negativ nu exclude complet diagnosticul, in cazuri rare alte tipuri de ciclina D putand fi supraexprimate2;5;13.
CD5 poate fi exprimat si in alte BLPC-B, in mod particular LPL sau SMZL, in multe din aceste cazuri expresia CD5 fiind parţiala. Expresia parţiala a CD5 nu exclude CLL sau MCL, iar pe de alta parte nu prezice un anume tip de limfom13. Posibile aspecte distinctive includ absenţa expresiei CD23 in majoritatea MZL si prezenţa diferenţierii plasmocitare intr-un subset semnificativ din acestea demonstrata de expresia CD138, expresia intensa a CD38 si restricţia lanţului usor de imunoglobuline citoplasmatice (cIg).
Totusi si alte subtipuri de neoplazii limfoide B pot demonstra diferenţiere plasmocitara, in special LPL2. De aceea, in cazul unei BLPC-B detectata prin IFCF a sangelui periferic sau maduvei osoase, cu expresia parţiala a CD5, pentru un diagnostic precis se recomanda biopsia tisulara2;13. Uneori studiile de genotipare pot susţine diagnosticul de MZL. Desi unele anomalii genetice pot fi intalnite atat in CLL/SLL cat si in MZL (de exemplu, trisomia 18), urmatoarele anomalii sunt mai tipice pentru MZL: t(11;18)(q21;q21), t(1;14)(p22;q32), t(14;18)(q32;q21), precum si deleţia 7q31 in SMZL2.
Un mic subset din pacienţii cu leucemie prolimfocitara B (B-PLL) pare sa fie CD5+. Unii dintre acestia au fost ulterior diagnosticaţi cu varianta blastoida de MCL prin testarea pentru translocaţia t(11;14)(q13;q32) sau rearanjamentele genei CCND1. Este dificil sa se distinga intre CLL cu numar crescut de prolimfocite, transformarea prolimfocitoida a CLL sau PLL de novo. Transformarea prolimfocitoida se caracterizeaza prin colorare scazuta pentru CD5, intensitate crescuta a colorarii pentru CD20 si sIg si achiziţia FMC-7. Totusi unii pacienţi cu transformare prolimfocitoida demonstreaza un fenotip identic cu cel al CLL/SLL precedente. Fenotipul B-PLL de novo este variabil, incluzand unii pacienţi CD5+, dar care, spre deosebire de CLL/SLL, de obicei sunt CD23– 2.
Este puţin probabil ca o BLPC cu celula mica B CD5 negativa sa reprezinte CLL, iar aceste cazuri sunt clasificate mai potrivit ca NHL in faza leucemica.
▪ Expresia CD10 la IFCF se coreleaza puternic cu diagnosticul tisular de limfom folicular (FL) sau limfom difuz cu celula mare B (DLBCL), urmate de limfomul Burkitt (BL)2;13. Expresia CD10 in alte tipuri de limfoame este neobisnuita, fiind foarte rara in LPL, MZL si MCL. Pe de alta parte CD10 este in mod normal exprimat de celulele B ale centrului folicular, leucemia/limfomul limfoblastic cu precursor de celula B, subseturi de celule T mature, limfoblasti precursori ce celula T, neutrofile si unele celule nonhematolimfoide.
Aproximativ 20-40% din toate DLBCL prezinta un fenotip CD10+, acestea putand fi dificil de diferenţiat de BL sau de FL compus din multe celule mari. Astfel cand un fenotip B matur CD10+ este identificat prin IFCF, distincţia intre aceste posibilitaţi trebuie evaluata in continuare prin morfologie2.
BL al copilului este in general CD10+ si CD5–. Desi BL al adultului este de obicei CD10+, fenotipul in aceste cazuri este mai variabil si este mai dificil de diferenţiat de DLBCL, dar spre deosebire de unii pacienţi cu DLBCL si majoritatea pacienţilor cu FL, BL este de obicei bcl-2– 2.
▪ HCL are un fenotip distinctiv care permite diagnosticul si detecţia unor niveluri scazute de boala ulterior terapiei: CD20 intens, CD22 intens, CD11c intens, CD25+, CD103+, sIg intermediar sau intens, FMC-7+, CD23–, CD5–, CD10–. Acest fenotip este mai sensibil si specific pentru diagnosticul de HCL decat coloraţia pentru fosfataza acida tartrat rezistenta (TRAP). CD103 este caracteristic exprimat in HCL, alte boli CD103+ fiind rare si nu raspund bine la acelasi tip de terapie, acestea incluzand HCL varianta (HCLv) si limfomul pulpei rosii a splinei cu limfocite viloase (SRPL), care sunt frecvent CD103+, precum si rare cazuri de SMZL si DLBCL.
Coexpresia CD103 si CD25 este considerata unul din cele mai utile criterii de diagnostic pentru HCL. Ocazional HCL clasica deviaza de la fenotipul caracteristic. Un mic subset de HCL (10-26%) sunt CD10+, dar sunt morfologic similare HCL CD10– si raspund la terapia tipica. Astfel diagnosticul de HCL trebuie luat in considerare atunci cand se identifica un fenotip CD10+, CD5– asociat cu identificarea altor aspecte fenotipice caracteristice HCL.
Alte variaţii fenotipice raportate includ lipsa CD103 (foarte rar), lipsa CD25, expresia bcl-1 sau prezenţa colorarii pentru CD23. Cazurile CD25– au morfologie variabila, de la HCLv si SMZL la PLL si DLBCL. Recent, coloraţia histochimica pentru anexina A1 a fost aratat ca distinge cu o sensibilitate si specificitate de 100% intre HCL si alte BLPC-B, incluzand SMZL si HCLv. HCLv se caracterizeaza prin numar mai mare de leucocite, absenţa monocitopeniei, prezenţa de celule cu nucleoli proeminenţi, lipsa colorarii pentru TRAP, CD25–, dar in rest este similara fenotipic HCL clasice. Este importanta recunoasterea acestei HCL variante deoarece raspunsul la agenţii eficienţi in HCL clasica este de obicei slaba, iar supravieţuirea mediana este semnificativ scurtata.
Totusi existenţa HCLv este dezbatuta si s-a pus intrebarea daca unele din aceste neoplasme nu reprezinta SMZL, cu care prezinta similitudini morfologice. In clasificarea OMS 2008, HCLv si SRPL au fost listate sub termenul de “limfom splenic cu celula B, neclasificabil”, HCLv nemaifiind considerata legata biologic de HCL2;4;5.
- MZL prezinta de obicei un fenotip CD5–, CD10–, iar diagnosticul se bazeaza pe identificarea trasaturilor morfologice caracteristice si excluderea altor neoplazii cu celula mica B: FL CD10–, MCL CD5– si HCL. Distincţia de HCL este mai dificila datorita fenotipurilor care se pot suprapune. MZL este adesea CD11c+ si poate fi pozitiv pentru CD103. Totusi in MZL colorarea pentru CD11c este mai slaba, lipseste combinaţia CD103, CD11c si CD25 si lipseste colorarea intensa pentru CD20 si CD222.
- Aproximativ 60-80% din pacienţii cu LPL sunt raportaţi cu un fenotip CD5–, CD10–, CD23–; mulţi pacienţi exprima CD11c si CD25, dar spre deosebire de HCL sunt CD103– 2.
▪ Neoplazii limfoide cu celula B matura care sa exprime atat CD5 cat si CD10 sunt puţin comune. Acest grup include, in ordinea incidenţei: DLBCL, FL, MCL, CLL/SLL, BL. Evaluarea morfologica si studii genetice (rearanjamentele genei BCL-2 in FL, tranlocaţia t(11;14)(q13;q32) sau rearanjamentele genei CCND1 in MCL, translocaţiile MYC in BL) pot fi necesare pentru stabilirea diagnosticului2.
Expresia CD38 si ZAP-70 a fost raportata ca ar avea valoare prognostica in CLL/SLL. Desi majoritatea studiilor au utilizat un cut-off de 30% celule pozitive pentru a determina pozitivitatea expresiei CD38, unele studii au demonstrat un prognostic advers pentru pacienţii cu expresia CD38 pe 20% sau chiar mai puţine celule tumorale2. Expresia ZAP-70 a fost aratat ca se coreleaza cu statusul mutaţional al regiunii variabile a lanţului greu de Ig (IgVH), progresie mai rapida a bolii si supravieţuire mai mica5.
Exista o serie de dificultaţi tehnice in determinarea statusului ZAP-70, legate de intensitatea relativ slaba a colorarii cu mulţi fluorocromi disponibili comercial, localizarea intracitoplasmatica a marker-ului care face necesara folosirea de tehnici de permealizare care pot duce la scaderea intensitaţii colorarii, prezenţa colorarii nespecifice la nivelul celulelor B nonneoplazice, expresia labila in timp si sensibilitatea la diferiţi anticoagulanţi (a fost recomandata utilizarea EDTA si transportul in maxim 24 ore in laborator).
Nu s-a ajuns la un consens in ceea ce priveste metoda optima de determinare a caror celule ar trebui considerate pozitive pentru ZAP-70. Recent au fost propuse cateva sisteme de calculare implicand media si mediana intensitaţii fluorescenţei ZAP-70 in celulele leucemice, celulele T, NK si celulele B normale2.
Neoplaziile limfoide cu celula T si NK matura
IFCF poate contribui la diagnosticul si clasificarea neoplaziilor limfoide cu celula T si NK matura, insa adesea celulele T sau NK anormale fenotipic sunt mai dificil de identificat in comparaţie cu celulele B mature anormale. Pe de alta parte clasificarea neoplaziilor cu celula T si NK este mai puţin bine stabilita decat cea a neoplaziilor cu celula B si multe neoplazii raman in categoria limfomului cu celula T periferica, nespecificat (PTCL,U). Clasificarea conform schemei curente nu urmeaza un algoritm simplu si necesita adesea asimilarea de informaţii diverse, incluzand studii morfologice, imunohistochimice, citometrie in flux, citogenetica, FISH si teste moleculare2.
Neoplaziile cu celula T/NK matura (periferica) sunt divizate in leucemice, cutanate si extraganglionare si cele cu origine ganglionara. Majoritatea limfoamelor cu celula T periferica (PTCL – PTCL, U, limfomul T angioimunoblastic, limfomul cu celula mare anaplazica) sunt malignitaţi posttimice, care exprima TCR α/β, derivate aparent din sistemul imun adaptativ (antigen specific bazat pe receptor). Ele apar in general in organele limfoide periferice din celule T naive, efectoare (reglatoare CD4+ si citotoxice CD8+) sau de memorie. Limfoamele cu celula NK si un numar mic de PTCL ce pot fi derivate extratimic sunt legate de sistemul imun innascut (nerestricţionat MHC). PTCL din acest grup (limfomul cu celula T hepatosplenic, limfomul cu celula T subcutanat panniculitis-like, limfomul cu celula T tip enteropatie) tind sa apara in localizari mucoase si cutanate, adesea exprima TCR γ/δ, antigene asociate celulei NK si proteine asociate granulelor citotoxice11.
Neoplasmele cu celula T si NK matura pot fi identificate prin IFCF pe baza detecţiei expresiei antigenice aberante, spre deosebire de celulele B neexistand un marker surogat de clonalitate la fel de fidel ca expresia kappa si lambda. In plus expresia antigenica aberanta trebuie distinsa de variaţiile fenotipice normale observate intre multiplele subseturi de celule nonneoplazice.
Ocazional expresia antigenica aberanta poate consta in lipsa completa a colorarii pentru unul sau mai multe antigene pan-celula T, CD5 si CD7 fiind antigenele cel mai frecvent pierdute. Celulele neoplazice CD7– trebuie distinse de o populaţie mica de celule T normale CD7– din piele si sange care pot creste in dermatoze benigne si alte condiţii reactive.
Mai frecvent neoplaziile T si NK demonstreaza o alterare mai subtila a colorarii pentru antigene decat lipsa completa a acestora, cele mai frecvente fiind colorarea celulelor T mai intensa sau mai slaba pentru CD3 sau CD5, colorarea mai slaba a celulelor NK pentru CD2, CD7, CD56 si CD57, o expresie intensa mai omogena a CD8 si CD16 de catre celulele NK.
De asemenea populaţiile de celule T si NK anormale pot fi recunoscute dupa expresia de antigene care nu sunt in mod normal exprimate in aceste linii, spre exemplu antigenele mieloide CD15, CD13 si CD33 au fost descrise in unele neoplazii cu celula T matura, putand duce la confuzia cu leucemia acuta mieloida; celulele NK pot achiziţiona CD5; expresia antigenului celulei B CD20 a fost descrisa intr-un procent mic de neoplazii cu celula T si poate fi de asemenea detectata intr-un subset mic de celule T normale.
Evaluarea prin IFCF a expresiei TCR V-β permite identificarea de populaţii restricţionate de celule T, dar, spre deosebire de testele PCR pentru rearanjamentele genei TCR care pot fi efectuate atat pe specimene proaspete cat si ingheţate si fixate, acest test este laborios si necesita analizarea, de preferinţa, in 24 ore de la recoltare.
Celulele NK nu prezinta expresia TCR, pentru demonstrarea clonalitaţii celulelor NK fiind dezvoltata analiza prin citometrie in flux a expresiei receptoruui NK (NKR), incluzand receptorii pentru Ig ai celulelor killer (KIR) si complexul CD94/NKG2, care pot fi de asemenea aplicate celulelor T citotoxice de memorie din leucemia cu limfocite mari granulare (LGL) cu celule T.
Informaţiile imunofenotipice formeaza o parte importanta a clasificarii neoplasmelor limfoide cu celula T si NK matura, acestea putand fi efectuate fie prin citometrie in flux, fie prin imunohistochimie (IHC), selectarea tehnicii optime depinzand de aspectul care este cercetat. Studiile de IFCF sunt superioare IHC in distincţia intre celulele T si NK. Anticorpii utilizaţi de IFCF detecteaza de obicei intregul complex TCR-CD3, prezent pe suprafaţa celulelor T si absent de pe suprafaţa celulelor NK, spre deosebire de IHC care detecteaza numai componenta epsilon a CD3, neputand distinge intre celulele T si NK. Celulele CD3– trebuie evaluate in continuare pentru diferenţierea intre celulele NK, celulele T anormale cu lipsa aberanta a CD3 si celulele T imature. Studiile moleculare care demonstreaza rearanjamentul clonal al genei TCR pot de asemenea confirma linia celulara T.
IFCF permite determinarea expresiei ambelor forme, α/β si γ/δ, ale TCR, spre deosebire de IHC care detecteaza numai forma α/β a receptorului, aceasta distinctie fiind importanta deoarece limfoamele cu celela T γ/δ sunt mai agresive.
Expresia CD4 si CD8 poate fi stabilita atat prin IFCF cat si prin IHC, IFCF prezentand avantajul identificarii celulelor neoplazice pe baza expresiei aberante a altor antigene si apoi izolarea lor pentru caracterizarea in continuare a acestora.
Atat IHC cat si IFCF pot fi utilizate pentru identificarea diferenţierii NK prin detecţia CD56 si CD57, IFCF fiind considerata o tehnica mai sensibila. In plus IFCF poate detecta CD16 si ar trebui utilizaţi mai degraba anticorpii pentru detecţia izoformei CD16A decat a izoformei 16B asociata granulocitelor.
Coloraţii pentru proteinele asociate granulelor citotoxice TIA-1, care identifica majoritatea celulelor T citotoxice, si granzima B sau perforina, care indica un fenotip citotoxic activat, pot fi efectuate atat prin IFCF, cat si prin IHC. De asemenea coloraţii pentru CD30 si proteina Alk-1 asociate cu limfomul cu celula mare anaplazica (ALCL) sunt mai frecvent efectuate prin IHC, dar sunt disponibile si prin IFCF.
CD103, exprimat de un subset de celule T intramucoase normale si in limfomul cu celula T asociat enteropatiei (EATCL), este detectabil prin IFCF2.
Neoplaziile cu ceula T matura
Expresia CD4 si CD8 poate fi utilizata pentru formularea unei liste de posibilitaţi diagnostice si determinarea informaţiilor necesare pentru subclasificarea acestora.
▪ Neoplaziile CD4+/CD8– includ sindromul Sézary/limfomul cu celula T cutanat (CTCL); leucemia prolimfocitara cu celula T (T-PLL); leucemia/limfomul cu celula T al adultului (ATLL); ALCL; limfomul cu celula T angioimunoblastic (AITCL); PTCL, U; rar LGL.
In primul rand vor fi luate in considerare bolile care afecteaza primar sangele, incluzand sindromul Sézary/CTCL, ATLL si T-PLL2.
- Sindromul Sézary/CTCL prezinta de obicei CD4+, CD26–, CD7–, cu prezenţa variabila a colorarii si intensitate variabila a CD25, acest fenotip nefiind specific bolii2;5.
- ATLL are un fenotip similar sindromului Sézary/CTCL CD4+, CD7–, dar cu o colorare intensa mai uniforma pentru markerul de activare CD25 (receptorul IL-2). De asemenea acesti pacienţi prezinta integrarea clonala a genomului HTLV-12;5;11 .
- T-PLL este cel mai frecvent CD4+ (60%), dar poate fi si CD4+, CD8+ (25%) si mai rar numai CD8+ (15%). Spre deosebire de CTCL si ATLL, T-PLL este de obicei CD7+, CD25–, lipseste pierderea aberanta sau scaderea expresiei altor antigene T si este de obicei negativ pentru antigene asociate NK si pentru proteinele asociate granulelor citotoxice. Diagnosticul trebuie sa ia in considerare excluderea cazurilor rare de LGL CD4+. Studiile citogenetice pot confirma diagnosticul, peste 80% din pacienţii cu T-PLL avand fie inv(14)(q11;q32), fie t(14;14)(q11;q32), implicand gena TCL1 si locusul TCR α/δ2,6.
- ALCL afecteaza primar ganglionii limfatici si pielea, dar in mod particular poate prezenta afectarea sangelui periferic. Aceasta entitate includea iniţial toate limfoamele compuse din celule mari care exprima uniform CD30 (Ki-1), insa clasificarea OMS curenta include acele cazuri care exprima un fenotip T sau nul, excluzand limfoamele cu celula mare B CD30+. ALCL este de obicei CD4+, poate exprima CD56, este cel mai frecvent CD2+, dar adesea lipsesc multe alte antigene T, incluzand CD3, CD5 si CD7 si poate exprima antigene mieloide CD13, CD15, CD33, putand duce la confuzia cu leucemia acuta mieloida. Un diagnostic definitiv poate fi stabilit la pacienţi care exprima proteina Alk-1 sau prezinta rearanjamentul genei ALK1 de pe cromozomul 2 la examenul citigenetic clasic sau FISH2,11. Rearanjamentele ALK sunt prezente la 50-70% din ALCL CD30+, t(2;5) aparand la aproximativ trei patrimi din acesti pacienţi. Expresia Alk subdivide ALCL in 3 subtipuri clinice: a) ALCL sistemic Alk+, b) ALCL sistemic Alk– si c) ALCL primar cutanat (de asemenea Alk–). ALCL Alk– poate fi considerat ca ALCL secundar cand urmeaza mycosis fungoides, papulozei limfomatoide sau bolii Hodgkin6.
AITCL este de obicei CD4+, CD2+, CD5+, prezinta adesea scaderea expresiei CD7 si uneori a CD3. Un subset de celule neoplazice pot fi CD10+, expresia CD10 de catre celulele T putand fi intalnita si in ALL cu precursor de celula T, rar in alte neoplasme cu celula T matura si intr-un subset de celule T normale. Aproape toţi pacienţii prezinta celule B EBV pozitive, iar aproximativ o treime prezinta de asemenea rearanjamente ale genei Ig2,6.
Asa cum este de asteptat, fenotipul PTCL,U este heterogen, neoplasmele din aceasta categorie fiind de obicei CD4+ si prezentand pierderea aberanta a expresiei CD7 si CD5. Prin definiţie, alte entitaţi specificate trebuie excluse inaintea stabilirii diagnosticului de PTCL,U2;5. Expresia aberanta a CD20 a fost raportata in PTCL,U, fiind propus ca aceste celule ar putea reprezenta contrapartea neoplazica a subpopulaţiei mici de celule T CD20+ prezente la persoanele sanatoase7.
▪ Cel mai frecvent neoplasm cu celula T CD4–/CD8+ este T-LGL. In sangele periferic alte entitaţi care trebuie luate in considerare sunt PTCL,U CD8+, o mica proporţie din PLL si rar CTCL/sindromul Sézary. In splina si alte situsuri extraganglionare diagnosticul diferenţial include si limfomul cu celula T subcutanat panniculitis-like (SPTCL), rar limfomul cu celula T hepatosplenic (HSTCL) si limfomul cu celula T γ/δ nonhepatosplenic.
- T-LGL prezinta un fenotip CD8+, CD3+, frecvent cu scaderea intensitaţii pentru CD5 si CD7 si expresia de antigene asociate NK, majoritatea pacienţilor exprimand CD57, mulţi CD16 si caţiva sunt CD56+. In plus, sunt exprimate de obicei proteinele asociate granulelor citotoxice TIA-1, granzima B si perforina. Desi majoritatea pacienţilor exprima TCR α/β, au fost raportaţi si pacienţi TCR γ/δ. LGL cu un fenotip NK poate prezenta de asemenea un fenotip CD8+, CD4–, dar lipseste expresia complexului CD3-TCR, astfel neintrand in categoria neoplaziilor limfoide cu celula T matura2,5,6.
- SPTCL este un limfom cu celula T citotoxica rar, de obicei CD8+, prezinta expresia uneia sau mai multor proteine asociate granulelor citotoxice, este numai focal pozitiv pentru CD56, este de obicei EBV– si cel mai frecvent TCR α/β2,11.
- HSTCL este de obicei CD4–, CD8–, rar fiind descrisi pacienţi CD8+, care pot fi dificil de diferenţiat de LGL. In comun cu T-LGL, HSTCL afecteaza de obicei splina si poate afecta sangele periferic, este frecvent CD16+ si exprima un fenotip citotoxic nonactivat cu prezenţa TIA-1 si absenţa granzimei B si perforinei, dar difera de majoritatea LGL prin expresia CD56 si lipsa CD57, demonstreaza absenţa completa a CD5 mai degraba decat scaderea intensitaţii acestuia, cel mai adesea exprima TCR γ/δ, exprima receptori de celula killer Ig-like (KIR), sugerand derivarea din celule T de memorie, si demonstreaza frecvent la examenul citogenetic isocromozomul 7q2,11.
- Cazuri rare de limfom cu celula T γ/δ nonhepatosplenic sunt CD8+, dar nu sunt confundate cu LGL deoarece lipseste afectarea splinei si a sangelui periferic2.
- O proporţie mica din CTCL sunt CD8+ si trebuie distinse de limfomul cu celula T γ/δ cutanat nonhepatosplenic mai agresiv. CTCL este de obicei negativ pentru proteinele asociate granulelor citotoxice, dar acestea pot fi prezente in transformarea cu celula mare a CTCL. Morfologia si aspectele clinice raman criterii importante de diferenţiere2.
▪ Expresia duala CD4+/CD8+ este neobisnuita in limfoamele cu celula T matura si trebuie sa duca la considerarea ALL cu precursor de celula T.
Cel mai caracteristic neoplasm limfoid cu celula T matura CD4+/CD8+ este T-PLL. Acesta exprima CD3 de suprafaţa si un set complet de antigene T ca CD2, CD5 si CD7, lipsesc markerii de imaturitate CD34, CD10 si TdT si demonstreaza inv(14)(q11;q32) sau t(14;14)(q11;q32).
Rar pacienţi cu ATLL, LGL si PTCL,U coexprima CD4 si CD82.
▪ Categoria CD4–/CD8– include EATCL, HSTCL, limfomul cu celula T γ/δ nonhepatosplenic, PTCL,U si rar limfomul cu celula NK/T tip nazal.
- EATCL este de obicei negativ pentru CD4, CD8 si CD5, exprima CD3 si CD7, TCR α/β+, este pozitiv pentru proteinele asociate granulelor citotoxice TIA-1, granzima B si perforina, este CD103+, poate fi CD56+ sau CD56– si poate exprima CD30, iar aceste cazuri trebuie diferenţiate de ALCL. Recent EATCL a fost asociat cu achiziţii in cromozomul 9q33-34 ce pot ajuta la confirmarea diagnosticului. Limfocitele intramucoase din boala celiaca pot demonstra un fenotip similar ca si rearanjamente clonale al genei TCR, diagnosticul de EATCL necesitand prezenţa unui infiltrat distructiv2,5,11.
- Limfomul cu celula T γ/δ nonhepatosplenic demonstreaza un fenotip similar HSTCL, dar de obicei cu un fenotip citotoxic activat: CD4–, CD8–, CD2+, CD3+, CD5–, C56+, in principal CD57–, TIA-1+, granzima B+, perforina+. Un numar semnificativ din limfoamele mucoase T γ/δ sunt asociate cu EBV, in timp ce cele cutanate sunt de obicei EBV– 2.
Neoplaziile cu celula NK matura includ limfomul extraganglionar cu celula T/NK tip nazal (EN-NK/T-NT), leucemia agresiva cu celula NK si un subset de LGL. Neoplaziile cu celula NK pot fi localizate sau diseminate in momentul examinarii iniţiale si majoritatea se comporta agresiv, dar este importanta diferenţierea LGL cu celula NK care prezinta un curs mai indolent. Spre deosebire de celelalte neoplazii cu celula NK aceasta exprima adesea CD57 alaturi de CD56 si este EBV–.
-
Limfomul extraganglionar cu celula T/NK tip nazal tipic este CD4–, CD8–, CD3s–, CD5–, CD2+, C56+, iar spre deosebire de NK normale este de obicei CD7–, CD16–. Celulele tumorale exprima lanţul ε al CD3 in citoplasma si astfel se coloreaza pozitiv pentru CD3 la IHC. Celulele exprima proteinele asociate granulelor citotoxice TIA-1, granzima B si perforina, iar EBV este prezent in forma epizomala clonala, respectiv EBER (EBV small encoded RNA) pozitiv, ceea ce implica rolul acestui virus in patogeneza. Cazurile de EN-NK/T-NT care se prezinta cu alte localizari decat cea nazala clasica (cel mai frecvent piele, tract gastrointestinal, testicul), inclusiv ganglionara, dar sunt in rest tipice, sunt incadrate in aceeasi categorie2;5;7. EN-NK/T-NT aparut la nivelul pielii trebuie diferenţiat de mycosis fungoides pleomorfic/transformat, PTCL,U si neoplaziile hematodermice CD4+, C56+, iar cel aparut la nivel intestinal trebuie diferenţiat de EATCL. Un subset bine recunoscut al EN-NK/T-NT are originea intr-o celula T citotoxica NK-like, acestea exprimand CD3 de suprafaţa si prezinta rearanjamente clonale ale genei TCR. Ramane problematica clasificarea cazurilor atipice in care lipseste expresia CD56 si proteinelor asociate granulelor citotoxice, precum si a formelor nonnazale in care lipseste dovada implicarii EBV, fiind sugerata incadrarea ca limfom cu celula NK neclasificabil. Clasificarea OMS a EN-NK/T-NT solicita atat pozitivitatea EBV, cat si a proteinelor asociate granulelor citotoxice7.
- Leucemia cu celula NK agresiva si limfomul extraganglionar cu celula T/NK tip nazal au un fenotip similar, diferenţierea fiind clinica, limfomul fiind localizat tisular, in timp ce leucemia se prezinta cu afectare medulara, citopenii si prezenţa de celule neoplazice circulante, simptome B, hepatosplenomegalie, teste funcţionale hepatice crescute, insoţite uneori de sindrom hemofagocitic2.
Neoplaziile blastice limfoide
includ leucemia acuta si limfomul limfoblastic. ALL si LL au caracteristici comune morfologice, imunofenotipice si citogenetice, distinctia dintre ele fiind considerata de multi arbitrara. Astfel in clasificarea OMS acestea sunt incadrate in categoria leucemie/limfom limfoblastic cu precursor de celula B si T. Desi unii pacienti cu LL pot aparea la evaluarea clinica si morfologica cu boala localizata, studii sensibile de IFCF si moleculare evidentiaza frecvent afectarea submicroscopica a maduvei osoase sau sangelui periferic14. IFCF permite identificarea celulelor imature sau anormale, distinctia lor de celulele imature prezente in mod normal in maduiva osoasa si timus, diferentierea intre ALL si AML, identificarea liniei celulare B sau T, subtiparea acestora si urmarirea raspunsului la tratament, incluzand identificarea responder-ilor precoce si detectia bolii minime reziduale2;5.
Utilizarea unui plot CD45 versus lumina dispersata lateral este foarte utila in identificarea blastilor, acestia prezentand o intensitate scazuta a expresiei CD45 si lumina dispersata lateral scazuta. Aceasta permite distinctia de limfocite (CD45 intens), precursorii eritroizi (CD45 negativ), precursorii neutrofilici si eozinofilici (lumina dispersata lateral mai crescuta) si monocite (lumina dispersata lateral mai crescuta si CD45 mai intens).
Populatia blastica este apoi izolata prin “gating” electronic pentru analiza detaliata a expresiei antigenice. Blastii neoplazici pot fi similari celulelor normale blocate in maturatie si sunt recunoscute la diagnosticul prin IFCF prin numarul crescut comparativ cu celulele normale care exprima acelasi fenotip dar si prin expresia frecventa de pattern-uri anormale de maturatie, denumite pattern-uri imunofenotipice asociate leucemiei, ce pot fi clasificate in 4 tipuri: 1) expresia aberanta de markeri care nu sunt in mod normal prezenti pe celulele liniei respective; 2) expresia asincrona sau fenotipul contine aspecte neasteptate pentru stadiul de maturatie respectiv, cum ar fi expresia CD20 pe suprafata precursorilor de celula B; 3) supraexpresia si 4) lipsa expresiei unor markeri1;2;5.
Distinctia intre AML si ALL este importanta pentru selectia terapiei corespunzatoare. In ALL CD19 are sensibilitatea si specificitatea cele mai mari pentru detectia liniei B, iar cCD3 pentru detectia liniei T. cCD22 sau cCD79a sunt de asemenea markeri sensibili si specifici pentru linia B2.
Tabel 17.9.5.4: Markeri pentru diagnosticul leucemiei acute limfoblastice8
|
Linia celulara |
Antigene |
|
ALL cu precursor de celula B |
CD19, CD10, CD79a, TdT, cCD22, HLA-DR, cCD79a |
|
ALL cu precursor de celula T |
CD1, CD2, CD3, CD4, CD5, CD7, CD8, TdT, cCD3 |
|
Leucemia Burkitt |
CD19, CD10, CD20, CD22, CD79a, sIgG, CD45 intens |
ALL demonstreaza frecvent expresia a 1 sau 2 antigene mieloide, care contribuie la detectia celulelor anormale si nu trebuie sa duca la diagnosticul de leucemie bifenotipica. Mai putin de 5% din cazuri prezinta heterogenitate lineala, fie leucemie bifenotipica (expresia de antigene de la mai mult de o linie de catre o singura populatie de blasti) sau leucemie bilineala (doua populatii de blasti din linii diferite).
Atunci cand o neoplazie blastica exprima markeri de la mai mult de o linie, celulele leucemice trebuie caracterizate complet morfologic, citochimic, imunofenotipic si citogenetic, iar informatiile trebuie cantarite si, de preferinta, stabilit un diagnostic de AML sau ALL. Aceeasi abordare este necesara la pacientii care exprima foarte putini markeri in incercarea de a minimiza numarul cazurilor din categoria AL nediferentiate2.
Este in dezbatere existenta AL de linie NK, parand a exista un grup denumit leucemie NK/mieloida, ce poate avea afectare extramedulara si poate exprima urmatorul imunofenotip: CD56+, CD7+, posibil CD2+, CD13+, CD33+, MPO– si CD3– (de suprafata si citoplasmatic)2.
Studiile de imunofenotipare au evidentiat diferente minore ale expresiei antigenice in LL cu precursor de celula B sau T si ALL. LL este rar la adult, dar este comun la copii. Aproximativ 90% din LL sunt de linie T, restul fiind de linie B, foarte rar putand avea originea intr-o celula NK. De obicei LL sunt TdT pozitive, un marker care distinge acest tip de neoplazie de alte tipuri de limfoame. Un numar mic de limfocite imature benigne TdT pozitive pot fi intalnite in sange si ganglionii limfatici.
CD34 este de asemenea exprimat de multe LL, fiind util pentru diferentierea acestora de limfomul Burkitt (BL). De asemenea LL cu precursor de celula T sau B poate exprima CD10 sau CALLA (common acute lymphoblastic leukemia antigen), dar expresia CD10 in T-LL nu a fost asociata cu un prognostic favorabil ca in T-ALL. LL cu precursor de celula B, ca si B-ALL, poate fi impartit mai departe in 4 subtipuri, pre-B precoce, pre-B, pre-B tardiv (tranzitional) si B matur, in functie de expresia lantului greu μ citoplasmatic si de suprafata si a lanturilor usoare de Ig. LL pre-B precoce exprima CD10, CD19, CD34 si TdT. Stadiul de maturatie pre-B este cel mai frecvent intalnit si exprima CD20 si lantul greu μ citoplasmatic fara Ig suprafata detectabile. Cazuri rare de LL cu precursor de celula B pot exprima Ig de suprafata fara TdT detectabil si acestea trebuie diferentiate de BL.
Tabel 17.9.5.5: Aspecte imunofenotipice in limfoamele limfoblastice cu precursor de celula B si T14
|
Subtip |
CD45 |
CD34 |
TdT |
CD3 |
CD5 |
CD7 |
CD19 |
CD20 |
CD22 |
CD79a |
CD10 |
Expresia Ig |
|
Pre-B precoce |
+ |
+ |
+ |
– |
– |
– |
+ |
+/- |
+ |
+ |
+ |
cIgμ–, sIgμ–, κ–, λ–
|
|
Pre-B |
+ |
+/- |
+ |
– |
– |
– |
+ |
+/- |
+ |
+ |
+ |
cIgμ+, sIgμ–, κ–, λ–
|
|
Pre-B tardiv (tranzitional) |
+ |
+/- |
+/- |
– |
– |
– |
+ |
+/- |
+ |
+ |
+ |
cIgμ+, sIgμ+, κ–, λ–
|
|
B matur |
+ |
+/- |
+/- |
– |
– |
– |
+ |
+/- |
+ | + | + |
cIgμ+, sIgμ+, κ±, λ±
|
|
T |
+ |
+/- |
+/- |
+ | +/- |
+ |
– |
– |
– |
– |
+/- |
Malignitatile limfoblastice cu precursor de celula T pot fi de asemenea subdivizate corespunzator cu stadiile de maturatie timica normala a celulelor T: precoce (CD7+, cCD3+, CD5+/-, CD2+/-, CD1a–, CD4–, CD8–), mijlociu (CD7+, CD5+, CD2+, cCD3+, sCD3+/-, CD1a+/-, CD4+/-, CD8+/-), si tardiv (CD7+, CD5+, CD2+, cCD3+, sCD3+, CD1a–, CD4+ sau CD8+). Majoritatea cazurilor de LL cu precursor de celula T corespund stadiului timic tardiv, ca si timomul, de care trebuie diferentiate. Majoritatea LL mentin expresia antigenelor pan-T, in timp ce in limfoamele cu celula mare T lipsesc unul sau mai multe din aceste antigene5;14. Expresia mai frecventa a TCR αβ decat γδ a fost raportata in T-LL fata de T-ALL. Unele LL pot prezenta expresia aberanta de antigene mieloide, iar T-LL pot exprima ocazional CD56. Toate recaderile T-LL trebuie restudiate imunologic deoarece un numar mic de cazuri pot prezenta o comutare fenotipica la leucemie acuta mieloida14.
ALL la adult sunt in 70% din cazuri cu precursor de celula B, 25% cu precursor de celula T si 5% cu celula B matura (celula Burkitt). Exista o incidenta usor mai mare a expresiei antigenelor mieloide in ALL la adulti decat la copii, comun fiind detectate CD13, CD15 si CD33. Cazurile rare cu coexpresia mai multor antigene mieloide si limfoide pot fi considerate leucemii acute bifenotipice.
Unele aspecte imunofenotipice prezinta valoare prognostica in ALL. Pacientii cu ALL cu celula B matura, caracterizata prin prin expresia lanturilor usoare de Ig de suprafata, κ sau λ, alaturi de alti markeri comuni ALL de linie B, incluzand CD10, CD19, CD20 si CD22, raspund prost la terapia standard, in aceste cazuri fiind stabilite programe de tratament cu doze intensificate ca terapie standard, ceea ce a dus la o imbunatatire semnificativa a supravietuirii la acesti pacienti.
De asemenea ALL cu celula T era considerata anterior o forma cu prognostic nefavorabil, dar cu programele de tratament moderne, imunofenotipul T a devenit cu prognostic favorabil. Prin analiza datelor imunofenotipice colectate prospectiv, grupul de studiu german CALGB a idetificat un subset, caracterizat prin expresia a mai putin de 3 markeri de celula T, cu prognostic nefavorabil. Blastii leucemici ai acestor pacienti exprima rar markeri de celula T matura, ca CD1, CD2, CD3, CD4 si CD8, corespunzand unui imunofenotip de ALL cu celula T precoce, de asemenea raportat ca nefavorabil.
Expresia CD34, mai frecventa la adult in ALL de linie B, a fost de asemenea raportata ca avand efect advers, dar aceasta se suprapune atat cu un numar crescut de leucocite, cat si cu prezenta cromozomului Ph, caracterizate de asemenea printr-un efect nefavorabil9.
La copii, ALL cu celula pre-B precoce cuprinde aproximativ doua treimi din cazuri, se asociaza cu un prognostic favorabil, majoritatea fiind CALLA pozitive, expresia acestuia neparand a avea semnificatie prognostica independenta15.
ALL cu celula pre-B reprezinta ~20% din cazuri. Expresia cIg se asociaza cu prezenta anomaliei citogenetice t(1;19)(q23;q13), identificata la 20-30% din pacientii cu ALL cu celula pre-B si asociata cu un prognostic advers la acesti pacienti. Restul pacientilor prezinta rate de supravietuire similare ALL cu celula pre-B precoce15.
ALL cu celula B matura este rara, cuprinzand 1-2% din cazurile de ALL la copii, la fel ca BL fiind caracterizata prin morfologie L3 FAB, un fenotip cu CD10, CD19, CD20, CD22, CD79a pozitive si TdT negativ, prezenta t(8;14) sau a unei translocatii inrudite si supraexpresia oncogenei c-MYC. Acesti copii prezinta varste mai mari, o incidenta mai mare a afectarii SNC si raspuns prost la tratamentul standard8;15.
ALL pre-B tranzitional, caracterizat prin expresia citoplasmatica si de suprafata a lanturilor grele fara expresia lanturilor usoare de Ig, apare la ~1% din copiii cu ALL, nu se asociaza cu morfologia L3 si cu translocatiile caracteristice ALL cu celula B matura si prezinta o evolutie favorabila15.
Unele fenotipuri in ALL se asociaza cu prezenta unor anomalii citogenetice si moleculare cu semnificatie prognostica. Astfel in B-ALL un fenotip CD9+, CD10+, CD19+, CD20– sau numai partial, CD34– este un marker sensibil pentru t(1;19)(q23;p13), dar este lipsit de specificitate1. Pe de alta parte un fenotip CD10–, CD15+, CD24– se asociaza cu t(4;11)(q21;q23), rearanjamente ale MLL si un prognostic prost2;5.
ALL cu celula T reprezinta ~15% din cazurile de ALL la copii, asociata anterior cu un prognostic nefavorabil, dar utilizarea regimurilor terapeutice intensive a rezultat intr-o supravietuire apropiata de cea a ALL non-T. Celulele leucemice pot fi dublu pozitive pentru CD4 si CD8. Majoritatea cazurilor de ALL-T sunt HLA-DR negative. Dintre acesti pacienti, cei cu imunofenotipul cel mai putin matur au o evolutie semnificativ mai proasta. Un subset caracterizat prin expresia CD7 si absenta CD4 si CD8 se asociaza cu rezistenta la chimioterapia conventionala, iar absenta expresiei CD2 identifica un grup de pacienti cu evolutie proasta.
Aproximativ 10-20% din copiii cu ALL exprima antigene mieloide, ca CD13 si CD33, care nu se asociaza cu semnificatie prognostica adversa conform ultimelor trial-uri, dar pot furniza un element convenabil pentru monitorizarea bolii minime reziduale8;15.
Expresia CD34 a fost asociata cu efect favorabil independent in ALL de linie B la copii, dar nu si in cea de linie T15.
Boala minima reziduala (MRD)
In prezent majoritatea testelor de citometrie in flux pentru MRD tintesc sa detecteze populatii reprezentand 0.01% din evenimente, respectiv o celula din 104, prin analiza a 500 mii pana la 1 milion celule, cu scopul detectarii a cel putin 50-100 evenimente de interes, putand concura astfel ca sensibilitate cu metodele PCR, citometria in flux prezentand avantajul discriminarii intre celulele viabile si cele moarte si masurarea directa a proportiei celulelor pozitive.
Provocarea pentru detectia MRD prin IFCF este separarea celulelor leucemice reziduale de celulele nonmaligne, mai ales in stadiile de regenerare a maduvei osoase in care aceasta poate contine mai multe forme de maturatie precoce. In 1997, Jennings si Foon au aratat necesitatea utilizarii unei prime “porti” CD45 versus lumina dispersata lateral (SSC) pentru diferentierea subseturilor de celule si, cel mai important, a celulelor imature/blastice cu SSC si expresia CD45 scazute, atat la diagnostic cat si pentru detectia MRD, printre celulele in maturare din maduva osoasa. In 1999, Campana si Coustan Smith au estimat ca primele 4 combinatii pentru detectia MRD in AL la IFCF sunt: 1) CD19/CD34/CD10; 2) CD13/CD33/CD34; 3) CD13/CD33/CD117; 1) CD13/CD34/CD117, ceea ce presupune ca aceste aspecte imunofenotipice trebuie urmarite la diagnostic.
Fenotipul se poate schimba adesea odata cu trecerea timpului si in urma tratamentului, astfel testul pentru MRD nu trebuie sa se bazeze pe potrivirea exacta a fenotipului bolii reziduale si cel al specimenului diagnostic original. Totusi subsetul de celule leucemice care persista mentin aspectul clonal si continua sa prezinte trasaturi omogene. Aceste celule reziduale apar la studiile prin IFCF ca un grup de celule ingust prezentand un fenotip “inghetat” printre celulele in maturare1;2.
Pentru leucemia acuta exista in general o diferenta de 1 log intre maduva osoasa si sangele periferic, cu nivelul cel mai mare de MRD in maduva osoasa. Astfel, ca si in studiile bazate pe tehnici moleculare, analiza maduvei osoase poate fi restrictionata la cazurile in care MRD este in mod repetat nedetectabila in sangele periferic2.
IFCF are un rol stabilit in detectia MRD in ALL, un rol in actualitate in CLL/SLL si un rol potential in alte malignitati hematopoietice si limfoide.
Cateva studii au demonstrat ca MRD detectata prin IFCF este un factor prognostic independent in ALL pediatrica. Astfel, prezenta MRD in maduva osoasa in absenta evidentei morfologice de boala, se asociaza cu risc mai mare de recadere, iar riscul creste cu nivelul de boala detectata. Schemele actuale de clasificare in ALL cu precursor de celula B incorporeaza, in afara criteriilor de risc NCI/Rome, elementele citogenetice, raspunsul precoce si incarcatura MRD la sfarsitul inductiei,.
La adult datele sunt mai putine, dar detectia MRD pare sa fie un factor de risc independent pentru recadere. Aceste informatii pot asista la identificarea unor pacienti cu risc crescut care pot beneficia de terapie aditionala. mai multe studii efectuate au aratat o buna corelatie intre datele obtinute prin IFCF si PCR1;2;15.
In ultimul timp, prin utilizarea schemelor de chimio-imunoterapie combinata, pot fi obtinute remisiuni MRD– in CLL/SLL, iar studiile initiale au aratat o durata a raspunsului si o supravietuire mai lungi la pacientii fara MRD detectabila. Studiile pentru MRD in CLL pot fi efectuate utilizand sange periferic. Totusi exista celule B normale ale sistemului imun innascut cu un fenotip asemanator celui al CLL-B. Pe de alta parte se cunoaste putin despre soarta acestor celule normale in urma chimioterapiei pentru CLL.
O procedura de citometrie in flux multiparametrica standardizata publicata recent de Rawstron et al a fost dezvoltata pentru detectia MRD la un nivel similar PCR-ului conventional (1×10-4 sau 0.01%). PCR-ul cantitativ cu oligonucleotide alela-specifice este mai sensibil, dar necesita generarea de primer-i specifici pentru pacient si nu este general disponibil, iar la un nivel de 0.01% se coreleaza cu rezultatele IFCF. Astfel IFCF multiparametrica standardizata a fost propusa ca metoda preferata pentru detectia MRD in CLL/SLL. Aceasta recomanda 3 combinatii, respectiv asocierea CD5/CD19 cu CD20/CD38, CD81/CD22 si CD79b/CD431;2.
Recomandari imunofenotipare sindroame limfoproliferative
- evaluarea limfocitozelor de etiologie neprecizata, la pacienti care prezinta adenopatii, transpiratii nocturne, febra, scadere ponderala, oboseala, infectii, anemie, tendinta la sangerare;
- distinctia intre limfocitozele policlonale (reactive, cel mai adesea virale) si limfocitozele monoclonale;
- identificarea afectiunilor limfoproliferative cu celula B sau T;
- subclasificarea fenotipica a bolilor limfoproliferative cronice;
- distinctia intre leucemia limfoblastica acuta (LAL) si leucemia mieloida acuta (LAM);
- subtiparea imunologica a LAL;
- distinctia dintre limfomul malign si leucemia acuta;
- monitorizarea tratamentului;
- depistarea precoce a recidivelor10,12.
Specimen recoltat – sange venos10.
Recipient de recoltare – vacutainer ce contine EDTA ca anticoagulant10.
Volum proba – 5 mL sange10.
Stabilitate proba – sangele trebuie sa ajunga in maxim 24 ore la laboratorul la care se efectueaza testul si in aceasta perioada se pastreaza la temperatura camerei. Este contraindicata refrigerarea probei10.
Cauze de respingere a probei – specimene care au depasit intervalul de stabilitate, probe refrigerate sau congelate10.
Metoda – citometrie in flux10.
Valori de referinta si comunicarea rezultatelor
Buletinul final va contine intervalele de referinta pentru subseturile limfocitare adecvate varstei pacientului impreuna cu o interpretare a rezultatelor obtinute10.
Limite si interferente
- Restrictia clasei lanturilor usoare de Ig a fost raportata rar in populatii de celule B reactive non-clonale, spre exemplu populatii ce prezinta restrictia lantului usor lambda au fost identificate in boala Castleman multicentrica. Astfel restrictia lantului usor de Ig nu este sinonima cu monoclonalitatea. De asemenea unele populatii care prezinta restrictia lantului usor si sunt monoclonale nu sunt neoplazice, identificate de exemplu in hiperplazia foliculara florida, incluzand-o pe cea observata la pacientii cu HIV. Astfel, rezultatele imunofenotiparii trebuie interpretate in conjunctie cu alte date clinice, morfologice si uneori genotipice2.
- La pacientii fara un diagnostic anterior de neoplazie limfoida identificarea unei populatii mici de celule B anormale fenotipic (mai putin de 5% din toate celulele analizate) nu trebuie sa duca la stabilirea unui diagnostic nou decat in corelatie cu datele clinice, morfologice sau alte modificari2;11.
- In AL, procentul celulelor imature determinate prin IFCF poate diferi de numarul blastilor pe frotiurile din aspiratul medular, aceasta putand avea diferite explicatii, cum ar fi: dilutia mai mare cu sange periferic decat partea utilizata pentru frotiuri, includerea unor precursori imaturi normali (hamatogonii) la numaratoarea manuala, utilizarea unui numarator diferit pentru determinarea procentului de blasti, la numaratoarea manuala acestia fiind determinati ca procent din toate celulele nucleate, in timp ce la IFCF acestia sunt determinati ca procent din toate celulele analizate sau din celulele noneritroide (etapa de liza utilizata pentru indepartarea celulelor rosii anucleate indeparteaza de asemenea un numar variabil de precursori eritroizi nucleati). De asemenea blastii pot fi dificil de recunoscut la numaratoarea manuala sau pot fi distrusi la efectuarea frotiurilor. In plus celulele mieloide hipogranulare pot avea o lumina dispersata lateral scazuta si pot intra in regiunea blastilor in plot-ul CD45 versus dispersia laterala2.
Bibliografie
Produsul a fost adăugat în coș
În plus, ai la dispoziție 30 de zile pentru a veni la recoltare.