înapoi la lista
Pancreatita ereditara (PRSS1)
Inchide
Analiza Pancreatita ereditara (PRSS1) a fost înlocuită cu Profilul genetic pancreatită ereditară (CASR, CFTR, CPA1, CTRC, PRSS1, SPINK1). Mai multe detalii aici.
Informatii generale Pancreatita ereditara (PRSS1)
Pancreatita ereditara, descrisa prima data de Comfort si Steinberg in anul 1952, este o afectiune ce se transmite autozomal dominant, cu o penetranta de 80%. Se caracterizeaza prin debut precoce cu episoade recurente de pancreatita acuta la copii, tineri si adulti, ce progreseaza spre forma cronica cu insuficienta exo- si endocrina. Boala apare de obicei la mai mult de doi membri ai unei familii sau la mai multe generatii ale aceleiasi familii. De precizat este faptul ca in aceste cazuri agentul etiologic care declanseaza episoadele de pancreatita acuta nu poate fi identificat.
Manifestarile clinice ale formei ereditare a pancreatitei variaza de la forme usoare cu evolutie favorabila pana la forme severe cu prognostic grav. Dupa descrierea corelatiei intre pancreatita ereditara si o mutatie la nivelul bratului lung al cromozomului 7, o serie de studii genetice au dus la evidentierea altor defecte responsabile de inhibarea activitatii tripsinogenului, reglarea functiei pancreatice si modularea procesului inflamator. Exista cel putin patru tipuri mutatii care determina aparitia pancreatitei ereditare:
- mutatii in gena care codifica tripsinogenul cationic (PRSS1);
- mutatiile genei care codifica inhibitorul serin-proteazic al tripsinei (SPINK1);
- mutatii la nivelul genei fibrozei chistice (CFTR);
- polimorfism la nivelul genelor implicate in reglarea raspunsului inflamator (TNF, IL-1, IL-10).
Sucul pancreatic contine 3 izoforme ale tripsinogenului ce au fost diferentiate pe baza mobilitatii lor electroforetice: tripsinogenul cationic (PRSS1), anionic (PRSS2) si mesotripsinogen. In mod normal, tripsinogenul cationic reprezinta aproximativ 66% din totalul tripsinogenului, in timp ce forma anionica doar o treime. Mesotripsinogenul este o specie minora, reprezentand mai putin de 5% din totalul tripsinogenului sau 0.5% din totalul proteinelor ce intra in alcatuirea sucului pancreatic3. Tripsinogenul este secretat in celulele acinare pancreatice. Este activat in tripsina la nivelul duodenului de catre enterokinaza sau o alta molecula de tripsina, care scindeaza tripsinogenul la capatul N-terminal si indeparteaza un lant peptidic scurt (peptidul de activare a tripsinogenului, TAP).
Tripsina activeaza apoi o cascada de precursori enzimatici. Pentru evitarea activarii tripsinei in pancreas exista o serie de mecanisme de protectie4. Tripsina este alcatuita din 2 domenii proteice globulare conectate printr-un lant lateral unic ce este denumit “bucla” de autoliza, situat opus fata de situsul activ. Molecula de tripsina contine de asemenea un “buzunar“ de legare a calciului localizat in apropierea lantului lateral; lantul prezinta un reziduu de arginina la aminoacidul R122 ce reprezinta tinta pentru o alta molecula de tripsina. Astfel, clivajul enzimatic al lantului lateral in pozitia R122 de catre o a doua molecula de tripsina produce inactivarea rapida a primei molecule de tripsina prin autoliza.
Analiza biochimica sprijina ideea ca Arg122 este un aminoacid important pentru autoliza si mutatii ale acestui aminoacid conduc la cresterea stabilitatii tripsinei. ”Bucla” de autoliza este flexibila, iar R122 ajunge in apropierea “buzunarului“ de legare a calciului; pe masura ce concentratia calciului creste, acesta patrunde in “buzunar“ si limiteaza expunerea situsului R122 la atacul enzimatic al unei alte molecule de tripsina. Din acest motiv calciul detine un rol important nu numai in secretia tripsinei ci si in stabilizarea moleculei, tripsina fiind susceptibila la autoliza rapida in celulele acinare atunci cand concentratia calciului este scazuta, dar protejata de autoliza dupa secretia activa in ductele pancreatice cand concentratia calciului este crescuta. Pancreatita acuta se dezvolta ca urmare a activarii intrapancreatice a tripsinogenului care la randul sau converteste toate proenzimele proteolitice in forme active, fapt ce determina in final autodigestia pancreasului.
In urma descrierii mutatiei de pe cromozomul 7, multe studii efectuate au aratat faptul ca la nivelul genei care codifica molecula de tripsinogen cationic exista de fapt mai multe mutatii care se pot asocia cu pancreatita ereditara, cele mai frecvent descrise fiind R122H si N29I. O alta molecula modificata evidentiata la unele familii cu boala ereditara este inhibitorul serin-proteazic descris de Kazal (serine protease inhibitor Kazal type1 – SPINK1), cu rol in inhibarea activitatii tripsinice intrapancreatice. Aceasta enzima poate inactiva 10-20% din tripsina activa. Mutatiile specifice responsabile pentru pancreatita ereditara au fost identificate in 1996, atunci cand s-a confirmat ca gena care determina boala se gaseste pe cromozomul 7 (7q35). Whitcomb si colaboratorii sai au demonstrat la acesti pacienti existenta unei mutatii in exonul 3 din gena care codifica tripsinogenul cationic (PRSS1). Aceasta mutatie, in care guanina (G) este substituita cu adenina (A) in codonul 117, determina inlocuirea argininei (CGC) cu histidina (CAF) si a fost denumita la inceput R117H. Mutatia elimina situsul initial de hidroliza ceea ce face ca tripsinogenul/tripsina sa fie rezistente la autoliza si inactivare permanenta. In acest fel, atunci cand tripsinogenul este activat in tripsina intrapancreatic, in cantitati care depasesc capacitatea inhibitorie a inactivatorului SPINK1, iar tripsina ramane activa datorita mutatiei R117H, aceasta din urma poate activa toate proenzimele, initiind autodigestia pancreatica3.
O a doua mutatie in gena tripsinogenului cationic – N21I – se caracterizeaza prin inlocuirea adeninei cu tiamina (T) in exonul 2, conducand la substitutia asparaginei (ACC) cu izoleucina (ATC). Mecanismul prin care mutatia N29I cauzeaza pancreatita este neclar. Caracterizarea biochimica a mutatiei N29I utilizand tripsinogen recombinat nu a evidentiat nici un efect asupra stabilitatii tripsinei sau tripsinogenului. S-a sugerat totusi (pe baza a 4 studii efectuate in doua laboratoare independente) ca mutatia N29I ar creste autoactivarea tripsinogenului, modificand legarea inhibitorului specific la tripsina sau prin afectarea inactivarii tripsinei, prin modificarea accesibilitatii situsului la hidroliza. Modificarea conformationala a moleculei de tripsinogen sustine prima ipoteza. Aceste doua mutatii (R117H si N21I) au fost identificate in familiile cu pancreatita ereditara din multe tari (Franta, Germania, Marea Britanie, Japonia si SUA).
De la descoperirea mutatiilor de la nivelul genei tripsinogenului cationic, un sistem nou de nomenclatura a mutatiilor genice umane a fost elaborat si acceptat. Astfel s-a schimbat numele mutatiilor comune, din R117H in R122H si din N21I in N29I. Din punct de vedere clinic, pacientii cu mutatia R122H au o evolutie a bolii mai severa si un debut la o varsta mai tanara in comparatie cu pacientii ce prezinta mutatia N29I. O mutatie mai putin frecventa este A16V (substitutia alaninei cu valina in pozitia 16), care a fost identificata initial la trei pacienti cu pancreatita idiopatica si un pacient cu pancreatita ereditara.
Mecanismul patogen prin care mutatia A16V cauzeaza pancreatita este speculativ si se refera la modificarea situsului de clivaj al peptidului semnal (TAP) implicat in procesarea intracelulara a tripsinogenului4. Mutatia A16V are penetranta scazuta si este inregistrata la pacienti fara istoric familial de pancreatita cronica ceea ce sugereaza ca mutatiile PRSS1 nu urmeaza in exclusivitate modelul de transmitere autozomal-dominant3. Mutatia N29T, descrisa pentru prima data de Pfützer et al. in 2002, se asociaza cu un fenotip similar cu cel determinat de mutatia R122H, caracterizat prin cresterea stabilitatii tripsinei si autoactivare. Deoarece autoactivarea crescuta a fost asociata cu mutatiile R122H, N29I si N29T, iar N29I nu are nici efect asupra stabilitatii tripsinei, concluzia logica a fost ca autoactivarea este mecanismul patogen comun de declansare a pancreatitei asociate cu mutatii PRSS11;4. Au mai fost descrise si alte mutatii PRSS1 a caror semnificatie ramane inca neclara: -28 delTCC, D22G, K23R, P36R, G83E, K92N etc. In fig.1 se poate observa harta mutatiilor de la nivelul genei care codifica tripsinogenul cationic (PRSS1)1.
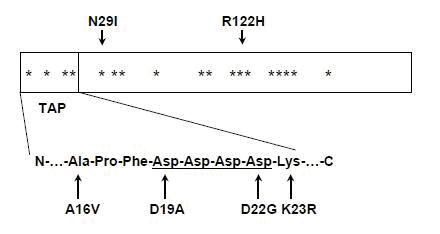

Fig.1 Harta lineara a mutatiilor asociate cu pancreatita ereditara in cadrul structurii primare a tripsinogenului cationic uman; pozitiile aminoacizilor afectati sunt marcati cu asterics; sunt indicate prin sageti pozitiile celor mai frecvente mutatii (R122H si N29I); este prezentata o parte din secventa TAP impreuna cu mutatiile intalnite in aceasta regiune; este subliniat motivul tetra-aspartat inalt conservat din cadrul moleculei TAP TAP – peptidul de activare a tripsinogenului (Adaptare dupa Hereditary chronic pancreatitis. In Orphanet Journal of Rare Diseases, 2:1, 2007.
Frecventa reala a mutatiilor PRSS1 la pacientii cu pancreatita idiopatica nu poate fi stabilita cu precizie. O metaanaliza indica o prevalenta medie de 1.9%, cu variatii intre 0.2 % si 10%3. Witt si colaboratorii au descris pentru prima data o asociatie intre mutatia genei ce codifica inhibitorul serin-proteazic Kazal tip 1 (SPINK1) si pancreatita cronica. SPINK1 este un inhibitor potent al tripsinei intrapancreatice. La incubarea unor cantitati echivalente de tripsina si SPINK1 se constata formarea unei legaturi covalente intre reziduurile catalitice de serina ale tripsinei si gruparea carboxil a lizinei din situsul reactiv al SPINK1.
Dupa incubare prelungita, activitatea tripsinei reapare in timp, lucru ce se explica prin faptul ca SPINK1 este degradata de tripsina. Gena SPINK1 este localizata pe bratul lung al cromozomului 5: 5q32; are o lungime de ~7.5 Kb si prezinta 4 exoni1;4. Mutatia cea mai frecvent observata la nivelul genei SPINK1 este N34S (o mutatie missense ce are la baza substitutia asparaginei cu serina la nivelul codonului 34). Se estimeaza ca 15-40% dintre pacientii cu pancreatita idiopatica poarta mutatia N34S pe una sau ambele alele. N34S se afla intr-un dezechilibru de linkaj complet cu alte 4 variante de secvente intronice: IVS-37T>C, IVS2+286A>G, IVS3-604G>A, IVS-66-65 insTTTT.
Au mai fost identificate si alte mutatii SPINK1: o mutatie homozigota a promotorului (-215G-A si 215 G-T), o mutatie la nivelul codonului start care distruge unicul codon de initiere a translatiei SPINK1 (M1T), precum si alte mutatii descrise doar la pacienti sau familii singulare. Spre deosebire de mutatiile PRSS1 care genereaza un „castig” functional, mutatiile SPINK1 cauzeaza o „pierdere” functionala ce se reflecta in scaderea inhibitiei proteazice. Mutatia N34S a fost asociata si cu pancreatita cronica alcoolica, precum si cu cea tropicala. Astfel, conform unui studiu, s-a inregistrat o frecventa de 5.8% la pacientii cu pancreatita alcoolica; de asemenea aceasta mutatie a fost depistata si la 20% dintre pacientii cu cu pancreatita tropicala calcificata si la 55% dintre pacientii diabet pancreatic fibrocalculos3. Deoarece 1-2% din pacientii din loturile control din studiile efectuate prezinta mutatia N34S, aceasta nu este suficienta pentru a explica patogeneza pancreatitei cronice la purtatorii mutatiei.
In plus, o analiza functionala cu inhibitor proteazic recombinant cu mutatie N34S a aratat o functie neschimbata a inactivatorului precum si o susceptibilitate nemodificata fata de tripsina. Aceasta indica faptul ca alte mecanisme decat schimbarea conformationala a N34S ar putea sta la baza predispozitiei de a dezvolta pancreatita cronica la purtatori. Manifestarile clinice ale pacientilor cu pancreatita ereditara se caracterizeaza prin episoade recurente de durere abdominala severa, care pot dura de la cateva zile la cateva saptamani. Nivelurile plasmatice ale amilazei si lipazei pot fi crescute pe parcursul episoadelor acute, dar in mod obisnuit sunt normale.
Evolutia clinica in timpul unui atac acut poate varia de la forma usoara (forma edematoasa) pana la cea severa, inflamatie necrotizanta a pancreasului. Din punct de vedere histopatologic pancreasul prezinta scleroza cu distrugere focala, segmentara sau difuza a parenchimului. Frecvent se pot observa dilatatii, stricturi sau obstructii in sistemul canalicular pancreatic. Initial, pancreatita cronica se caracterizeaza printr-o etapa de episoade recurente de pancreatita acuta (stadiu incipient), apoi se trece catre disfunctie progresiva pancreatica cu sau fara prezenta calcificarilor (stadiu avansat). Acesti pacienti pot dezvolta deseori calcificari pancreatice, diabet zaharat si steatoree si, in plus, au un risc ridicat de carcinom pancreatic. Pentru ameliorarea durerii se poate realiza uneori decompresie ductala. Aparitia unor simptome asemanatoare la rudele pacientilor cu pancreatita trebuie sa ridice suspiciunea unei boli pancreatice ereditare1;3;4.
De mentionat este faptul ca la copii simptomul cardinal este aparitia unei dureri epigastrice bruste si recurente. Spre deosebire de adulti, durerea prelungita nu este o constanta in manifestarile clinice comune la copii. De asemenea pot aparea greata, varsaturi si presiune abdominala. Unii copiii dezvolta insuficienta pancreatica cu steatoree si diabet zaharat insulinodependent1;3.
Recomandari pentru testarea genetica
Testarea genetica este indicata la pacientii cu:
- doua sau mai multe atacuri separate de pancreatita acuta de etiologie neprecizata;
- pancreatita cronica idiopatica;
- istoric familial de pancreatita acuta de etiologie neprecizatã la rudele de gradul I sau II;
- pancreatita acuta de etiologie neprecizata la copil, care necesita spitalizare1;3.
Diagnosticul de certitudine se stabileste in cazul pancreatitei ereditare prin secventiere genica. Cele mai multe laboratoare si-au concentrat testele asupra exonilor 2 si 3 din gena PRSS1, care au fost asociate pana in prezent fara ambiguitate cu pancreatitele determinate genetic. Cu toate acestea, este posibil ca noi mutatii sa fie identificate in regiunea exonilor 1, 4 si 5, in regiunea intronica sau promotorului. De asemenea triplicarea unui segment continand gena PRSS1 a fost evidentiata la anumiti pacienti cu pancreatita cronica1. Testarea persoanelor asimptomatice cu risc de boala implica, de obicei, interviuri pre-test, in care sunt solicitate motivele testarii, sunt discutate cunostintele individuale ale pacientului despre forma cu debut precoce si posibilul impact al rezultatelor testelor pozitive sau negative. Cei care doresc testarea ar trebui sa fie consiliati cu privire la eventualele probleme pe care le pot intampina legate de sanatate, invaliditate, educatie, discriminare, interactiune sociala si familiala.
De mentionat este faptul ca testele genetice la copii sunt o problema complexa, deoarece, in functie de varsta copilului, acesta nu poate fi intotdeauna inclus in procesul de luare a deciziei de testare genetica. Prin urmare consilierea genetica extinsa este necesara. Un alt aspect important il reprezinta faptul ca astfel de teste nu sunt utile pentru estimarea varstei de debut, severitatii, tipului de simptome sau modalitatii de progresie a bolii. Deoarece penetranta mutatiilor genei PRSS1 este incompleta si manifestarile clinice ale bolii sunt variabile in majoritatea familiilor, diagnosticul prenatal nu ar trebui incurajat. Chiar si in studiile recent publicate cu privire la testarea genetica in pancreatita ereditara autorii au avut rezerve in ceea ce priveste diagnosticul prenatal, dar subliniaza ca acesta nu poate fi refuzat unui pacient care este informat si a consimtit. Parintii care solicita acest test ar trebui informati ca in majoritatea cazurilor o evolutie dureroasa a bolii este autolimitata la cativa ani.
Diagnosticul prenatal la gravidele cu risc crescut pentru o mutatie PRSS1 este posibil prin analiza ADN-ul extras din celule fetale obtinute prin amniocenteza, efectuata de obicei la aproximativ 15-18 saptamani de gestatie sau biopsia vilozitatilor coriale, la aproximativ 10-12 saptamani de gestatie. Mutatiile cauzatoare de boala trebuie sa fie identificate inainte de testarea prenatala la un membru afectat al familiei1;3;4.
Specimen recoltat – sange venos2.
Recipient de recoltare – vacutainer ce contine EDTA ca anticoagulant2.
Cantitate recoltata – 5 mL sange2.
Cauze de respingere a probei – folosirea heparinei ca anticoagulant; probe coagulate sau hemolizate2.
Stabilitate proba – 7 zile la 2-8ºC2.
Metoda – secventierea tuturor exonilor PRSS1 si SPINK1 + analiza deletiilor/duplicatiilor MLPA2.
Raportarea si interpretarea rezultatelor
Un rezultat negativ este definit prin absenta mutatiilor in genele SPINK1 si PRSS1. Mutatiile R122H, N29I si A16V sunt responsabile de peste 90% din mutatiile PRSS1. Persoanele cu un rezultat pozitiv pentru mutatiile majore trebuie consiliate cu privire la modul de transmitere autozomal-dominant, penetranta incompleta, evolutia clinica variabila si strategiile de preventie a episoadelor de pancreatita acuta evitand factorii de risc asociati: alcool, medicamente, tulburari metabolice. Pacientii cu fenotipul de pancreatita ereditara trebuie evaluati radiologic si endoscopic pentru a identifica si trata factorii de risc, in special litiaza coledociana si alti factori obstructivi, care pot contribui la declansarea atacurilor de pancreatita acuta.
Avand in vedere riscul de cancer pancreatic la pacientii purtatori ai mutatiilor R122H sau N29I acestia trebuie sa fie consiliati sa renunte la fumat. Diferitele mutatii SPINK1 pot conduce la modele de transmitere variabile. Deoarece mutatia M1T distruge codonul start si genereaza o alela nula, aceasta se transmite autozomal-dominant.
Pe de alta parte mutatia N34S care reduce capacitatea SPINK1 da nastere unei tare recesive sau complexe. Astfel, in stare homozigota aceasta mutatie poate determina pancreatita, in timp ce la heterozigoti poate cauza o reducere subliminala a SPINK1, necesitand factori exogeni sau endogeni pentru a declansa manifestarile clinice3.
Limite si interferente
Faptul ca testele sunt negative pentru mutatiile tripsinogenului cationic sau SPINK1 nu exclude diagnosticul de pancreatita ereditara, fiind cunoscut faptul ca 30-40% din pacientii cu pancreatita ereditara sunt negativi pentru aceste mutatii1;3;4.
Bibliografie
- Jonas Rosendahl, Hans Bödeker, Joachim Mössner, Niels Teich. Hereditary chronic pancreatitis. In Orphanet Journal of Rare Diseases, 2:1, 2007.
- Laborator Synevo. Referintele specifice tehnologiei de lucru utilizate 2010. Ref Type: Catalog.
- ircea Grigorescu, Mircea Dan Grigorescu. Genetic Factors in Pancreatitis. In Romanian Journal of Gastroenterology, 2005, 14(1):53 – 61
- Richard M Charnley, Hereditary pancreatitis. In World J Gastroenterology 9(1):1-4, 2003.
Vezi tot conținutul
Vezi mai puțin
