înapoi la lista
Galactozemie - mutații GALT
Inchide
Informaţii generale - Galactozemia
Galactozemia este o boala cu transmitere autosomal recesiva, caracterizata prin perturbarea metabolismului galactozei cu aparitia unor complicatii ce pot pune viata nou-nascutului in pericol5.
Galactoza este o componenta a lactozei, glicolipidelor, glicoproteinelor si proteoglicanilor; metabolizarea acesteia realizandu-se cu precadere la nivelul ficatului si rinichiului. Conversia galactozei la glucoza implica urmatoarele etape (vezi figura 1):
- fosforilarea galactozei cu formarea galactozo-1-fosfatului, reactie ce se realizeaza cu ajutorul galactokinazei;
- transferul restului galactozil pe UDP-glucoza sub actiunea UDP-glucozo-galactozo-1-fosfat-uridil-transferaza (GALT);
- interconversia intre glucoza si galactoza are loc la nivelul nucleozid-difosfatilor, presupune inversarea configuratiei stereochimice a gruparii 4’ hidroxil si este catalizata de UDP-glucozo-4-epimeraza; aceasta etapa contribuie la regenerarea UDP-glucozei si astfel asigura continuitatea ciclului de metabolizare a galactozei.
Glucozo-1-fosfatul generat poate fi izomerizat in glucozo-6-fosfat care detine un rol important in procesul de glicoliza si in biosinteza inozitolului.
Galactoza poate fi transformata in galactitol sub actiunea aldoz-reductazei, in prezenta NADPH (sau NADH), aceasta reprezentand o cale alternativa de metabolizare a galactozei la pacientii cu galactozemie. De asemenea poate fi oxidata sub actiunea galactozo-dehidrogenazei, conducand la formarea acidului galactonic si CO21.
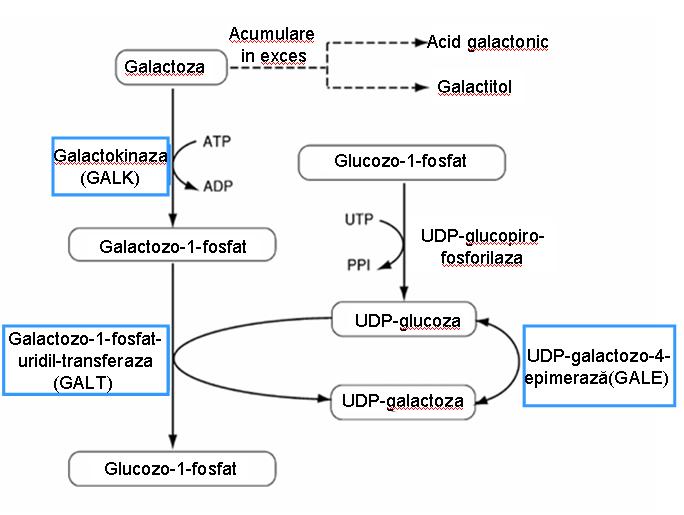

Fig. 1 Metabolismul galactozei (calea Leloir)
Hipergalactozemia este asociata cu 3 deficiente enzimatice:
- tipul I (GALT) – modificarea activitatii enzimatice a galactozo-1-fosfat-uridil-transferazei (GALT), ce este responsabila pentru galactozemia ereditara si este cel mai comun deficit enzimatic;
- tipul II (GALK1) – modificarea activitatii enzimatice a galactokinazei, ce converteste galactoza la galactozo-1-fosfat; este o deficienta rara;
- tipul III (GALE) – modificarea activitatii enzimatice a uridin-difosfat-glucozo-4-epimerazei, care epimerizeaza UDP galactoza la UDP glucoza si este, de asemenea, putin frecventa3.
| Tipul | Gena | Locus | Enzima | Nume |
| 1 | GALT | 9p13 | galactozo-1-fosfat-uridil-transferaza | Galactozemia clasica |
| 2 | GALK1 | 17q24 | galactokinaza | Deficienta de galactokinaza |
| 3 | GALE | 1p36-p35 | UDP-galactozo-4-epimeraza | Deficienta de UDP-galactozo-4-epimeraza |
Forma clasica (tipul I) a bolii si de altfel si cea mai severa este determinata de mutatii ale genei care codifica enzima galactozo-1-fosfat-uridil-transferaza (GALT). Este cea mai frecventa (1/48000 nou-nascuti) afectiune monogenica care intereseaza metabolismul carbohidratilor. Homozigotii pentru alela galactozemiei clasice (genotip G/G) au o activitate enzimatica scazuta sub 5%, ceea ce impiedica conversia eficienta a galactozei in glucoza.
Deficienta GALT duce la acumulare de galactozo-1-fosfat si galactoza in tesuturi, in special ficat, creier sau rinichi. Se crede ca afectarea hepatica (ciroza) si retardul mental induse de galactozemia clasica depind de cantitatea de galactozo-1-fosfat depusa in aceste tesuturi. Galactoza este convertita la galactitol in celule si produce efecte osmotice, cum ar fi edematierea fibrelor cristalinului ce poate conduce la cataracta si a neuronilor, care poate determina pseudotumor cerebri.
Ca si in forma asociata cu deficienta galactokinazei, cataracta se dezvolta secundar acumularii de galactitol la nivelul cristalinului. La femeile homozigote riscul de a dezvolta hipogonadism hipergonadotrop cu aparitia insuficientei ovariene este crescut si se manifesta la o varsta frageda chiar daca tratamentul dietetic este instituit prompt. Cu toate acestea, femeile afectate pot avea copii.
Heterozigotii (genotip G/N, N fiind alela normala) sunt asimptomatici, prezentand o scadere la jumatate a activitatii enzimei fata de persoanele ce nu prezinta mutatii3;5.
Gena ce codifica enzima GALT este situata pe bratul scurt al cromozomului 9 si este compusa din 11 exoni (vezi figura 2). Au fost identificate peste 180 mutatiii ale genei; un numar de 8 mutatii cauzatoare de boala (G) sunt intalnite in mod obisnuit la pacientii cu galactozemie clasica. Cea mai frecventa este mutatia punctiforma la nivelul exonului 6 care determina substitutia glutaminei cu arginina (Q188R; p.Gln188Arg) si care este responsabila de 60-70% din cazurile de boala la caucazieni.
Mutatia S135L (p.Ser135Leu) care determina substitutia serinei cu leucina este cel mai frecvent intalnita la populatia de rasa neagra fiind asociata cu peste 50% din cazurile de boala la aceasta categorie de persoane. Mutatia K285N (p.Lys285Asn) care determina substitutia lizinei cu asparagina in exonul 9 ocupa locul doi ca frecventa in cadrul alelelor asociate cu boala la caucazieni (28%). Mutatia L195P (p.Leu195Pro) care inlocuieste leucina cu prolina este inregistrata in 5-7% din cazurile de galactozemie clasica6;7.
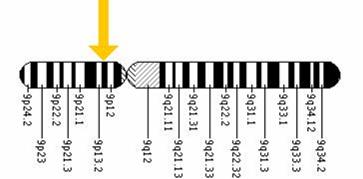
Fig. 2 Localizarea genei GALT pe cromozomul 9
(www.wiki.medpedia.com/Galactoso-1-phosphate uridylyltransferase)
In plus fata de forma clasica, exista varianta Duarte a galactozemiei – o conditie asimptomatica sau insotita de manifestari clinice usoare – ce rezulta ca urmare a afectarii partiale a enzimei GALT. Aceasta varianta se caracterizeaza printr-o heterozigotie compusa pentru o alela clasica G ce determina scaderea marcata GALT si o alela Duarte D-2 (D2; uneori denumita D) ce induce o afectare GALT partiala. Hemolizatele provenite de la pacienti cu galactozemie GALT demonstreaza in medie o activitate enzimatica de ~ 25%, desi sunt descrise variatii individuale mari. La electroforeza cu focalizare izoelectrica se obtine un aspect caracteristic de mobilitate alterata a izoenzimei GALT.
Studiile anterioare au indicat faptul ca alelele D sunt purtatoare ale unei mutatii missens p.N314D (c.940A>G) ce determina inlocuirea in pozitia 314 a asparaginei cu acidul aspartic. Fenotipurile Duarte (D/N, D/D si D/G) prezinta aproximativ 75, 50 si respectiv 25% din activitatea normala a GALT si apar cu o prevalenta de aproximativ 6% in populatia alba. Substitutia N314D este complet responsabila de alterarea mobilitatii enzimatice insa nu explica afectarea partiala a activitatii GALT.
Mai mult, aceasta mutatie se regaseste si pe alelele Duarte-1 (D1, denumite de asemenea Los Angeles sau LA) care asociaza o activitate enzimatica normala sau crescuta, fiind considerata din acest motiv o varianta comuna sau un polimorfism al genei GALT. Studii ulterioare ale alelelor D1 si D2 au aratat ca N314D se gaseste intr-un dezechilibru de linkaj cu alte variante de secvente nucleotidice, iar acestea difera intre D1 si D2.
Astfel, alelele D1 prezinta in mod specific o schimbare de nucleotide c.652C>T ce determina o substitutie silentioasa la nivelul codonului 218 (CTA →TTA, p.L218, denumita uneori si L218L), in timp ce alelele D2 prezinta o deletie 5’ de 4 bp a promotorului (c.-119_-116delGTCA) alaturi de 3 modificari de baze intronice. Ambele alele D1 si D2 poarta de asemenea o secventa extinsa de nucleotide A la nivelul intronului 10. Diferenta intre cele 2 alele in ceea ce priveste activitatea enzimatica GALT a ramas pentru mult timp un subiect de controversa. Un studiu recent indica existenta unei expresii reduse a ARNm la nivelul alelei D2 care contribuie la compromiterea functiei GALT; mai mult, deletia 5’ de 4 bp a promotorului ar reprezenta o mutatie cauzala in galactozemia Duarte2.
Deficienta galactokinazei (tipul II) poate fi suspectata la persoanele care au cataracta, galactozemie si cresterea excretiei urinare a galactitolului, dar nu prezinta alte manifestari clinice.
Prevalenta deficitului de GALK este necunoscuta, dar este probabil mai mica 1:100000. Detectarea unui nivel redus de activitate al galactozokinazei este un element important pentru stabilirea diagnosticului, dar confirmarea se face prin evidentierea mutatiilor genei care codifica enzima.
Poate fi limitata doar la nivelul eritrocitelor si leucocitelor si se asociaza cu dezvoltarea cataractei, dar nu determina retard in crestere, retard mintal sau afectare hepatica.
Administrarea de lactoza la femeile insarcinate cu deficit de galactokinaza poate determina aparitia cataractei fetale5.
Deficitul de UDP-galactozo-4-epimeraza (GALE) (tipul III) trebuie suspectat la persoanele care prezinta afectiuni hepatice, surditate neurosenzoriala asociate cu valori crescute ale galactozo-1-fosfatului eritrocitar si niveluri normale ale GALT. Diagnosticul se stabileste pe baza detectarii unui nivel scazut de activitate al UDP-galactozo-4-epimerazei si se confirma prin evidentierea mutatiilor genei care codifica enzima. Deficienta UDP-galactozo-4-epimerazei are o prevalenta estimata de 1:23000 in Japonia si este necunoscuta in alte populatii3;5.
Manifestarile clinice ale galactozemiei clasice apar la sugar, la cateva zile sau saptamani de la nastere si sunt determinate de ingestia laptelui matern sau a diverselor formule de lapte ce contin lactoza. Acestea constau in incapacitate de inghitire si voma, lipsa cresterii, leziuni hepatocelulare (hepatomegalie cu insuficienta hepatica), icter, hipoglicemie, hiperamoniemie, sangerare si sepsis. Retardul mental devine evident dupa 6-12 luni si este, de cele mai multe ori ireversibil.
Nou-nascutii cu forma clasica de galactozemie sunt susceptibili la infectii bacteriene generalizate (in special cu Escherichia coli) care pot cauza soc si moarte in perioada neonatala. Daca insa este instituita precoce o dieta fara lactoza simptomele se remit rapid si complicatiile (insuficienta hepatica, sepsisul, decesul neonatal) pot fi prevenite. Cataracta nu apare in mod obisnuit la nastere, dar se dezvolta treptat, fiind vizibila in termen de cateva saptamani sau luni. In ciuda tratamentului adecvat, copii cu galactozemie prezinta pe termen lung un risc crescut de dezvoltare a retardului de crestere si mental, a problemelor de vorbire (dispraxie verbala), cirozei hepatice macronodulare si insuficientei ovariane la femei. Prognosticul este in general rezervat si variabil la pacienti diferiti3;5;6.
Galactozemia este o afectiune care poate fi depistata prin screening neonatal. Clinitestul este cel mai simplu test screening si se bazeaza pe determinarea galactozei intr-o proba de urina. Acesta se efectueaza dupa 24-36 ore de la administrarea unui preparat ce contine lactoza. In unele tari se foloseste testul dintr-o picatura de sange recoltata pe hartie de filtru standardizata (card Guthrie) prin care se determina galactoza totala (galactoza si galactozo-1-fosfat) si/sau activitatea enzimei GALT. La copiii cu risc crescut se poate determina galactozo-1-fosfat din sangele recoltat din cordonul ombilical. Un test de screening ideal este acela care masoara activitatea GALT3;5.
Recomandari pentru testarea genetica Galactozemie - mutații GALT
Testarea genetica in vederea depistarii mutatiilor GALT este utilizata pentru confirmarea diagnosticului de galactozemie la copiii cu screening neonatal pozitiv. De asemenea este utila pentru diferentierea alelelor Duarte D2 de cele LA.
Astfel, testarea genetica defineste genotipul si permite stabilirea prognosticului.
Testarea rudelor apropiate pentru depistarea statusului de purtator asimptomatic se poate efectua in urma sfatului genetic si numai dupa identificarea mutatiilor cauzatoare de boala in familie. Intrucat galactozemia este transmisa autozomal recesiv parintii unui copil cu galactozemie clasica sunt obligatoriu heterozigoti pentru o alela mutanta G, iar fratii asimptomatici ai acestuia au un risc de 2/3 de a fi purtatori de alela G. De asemenea fratii parintilor au un risc de 50% de a fi purtatori.
Fratii unui copil cu galactozemie Duarte au in momentul conceptiei un risc de de 25% de galactozemie D/G daca parintii au genotipurile D/N si G/N si un risc de 25% de galactozemie clasica G/G daca parintii prezinta genotipurile D/G si G/N; din acest motiv este justificata testarea genetica a parintilor.
Diagnosticul prenatal pentru fetusii care prezinta un risc de 25% de galactozemie clasica (G/G) este posibil prin analiza ADN-ul extras din celule fetale obtinute prin amniocenteza, de obicei efectuata la aproximativ 15-18 saptamani de gestatie sau biopsia vilozitatilor coriale, la aproximativ 10-12 saptamani de gestatie. Si in aceste cazuri trebuie identificate in prealabil alele G din familie5.
Specimen recoltat – sange venos4.
Recipient de recoltare – vacutainer ce contine EDTA ca anticoagulant4.
Cantitate recoltata – 5 mL sange4.
Cauze de respingere a probei – folosirea heparinei ca anticoagulant; probe coagulate sau hemolizate4.
Stabilitate proba – 7 zile la 2-8ºC4.
Metoda – secventiere completa a genei GALT + analiza deletiilor/duplicatiilor MLPA; pot fi astfel depistate atat mutatiile asociate cu galactozemia clasica cat si variantele Duarte1.
Raportarea si interpretarea rezultatelor
Vor fi comunicate mutatiile depistate in gena GALT si genotipul respectiv4.
Bibliografie
1. David L. Nelson, Michael M. Cox. Glycolysis, Gluconeogenesis, and the Pentose Phosphate Pathway. In Lehninger Principles of Biochemistry, fifth edition, 2008, W. H. Freeman and Company, 545-546.2. Amanda E. Carney, Rebecca D. Sanders, Kerry R. Garza, Lee Ann McGaha, Lora J. H. Bean, Bradford W. Coffee, James W. Thomas, David J. Cutler, Natalie L. Kurtkaya, Judith L. Fridovich-Keil. Origins, distribution and expression of the Duarte-2 (D2) allele of galactose-1-phosphate uridylyltransferase. In Human Molecular Genetics, 2009, Vol. 18, No.9, 1624-1632.3. Gerard T Berry, George A Anadiotis, Galactose-1-Phosphate Uridyltransferase Deficiency (Galactosemia) www.emedicine.medscape.com, Ref Type: Internet Communication.4. Laborator Synevo. Referintele specifice tehnologiei de lucru utilizate 2010. Ref Type: Catalog.5. Louis J Elsas II. Galactosemia. Gene Reviews, 2000, www.ncbi.nlm.nih.gov. Reference Type: Internet Communication.6. Mayo Clinic, Mayo Medical Laboratories. Reference Laboratory Services for Health Care Organizations. Galactosemia Gene Analysis, Known Mutation. www.mayomedicallaboratories.com. 2010. Ref Type: Internet Communication.7. OMIM (Online Mendelian Inheritance in Man). Galactose-1-Phosphate Urydylyltransferase. https://www.ncbi.nlm.nih.gov. Reference Type: Internet Communication.8. Valeriu Popescu, Alis Antrasian, Andrei Zamfirescu. Screeningul neonatal in bolile genetice de metabolism. In Revista Romana de Pediatrie, volum LVIII, nr.4, 2009.Vezi tot conținutul
Vezi mai puțin