înapoi la lista
Adventia - Panel genetic comprehensiv pentru identificarea statusului de purtător (carrier screening)
Inchide
Informații generale Adventia - Panel genetic comprehensiv pentru identificarea statusului de purtător (carrier screening)
Adventia - Panel genetic comprehensiv pentru identificarea statusului de purtător (carrier screening) este un test genetic de ultimă generație destinat determinării statusului de purtător sănătos al unei variante asociate unei boli genetice la persoanele clinic sănătoase (fenotipic sănătoase). Informațiile genetice furnizate de Adventia pot contribui la informarea, orientarea și sprijinirea persoanelor în luarea deciziilor reproductive și la reducerea riscului ca purtătorii unor mutații genetice să transmită o boală genetică copiilor lor.
Condiții de recoltare:
Testarea va fi efectuată numai după consimțământul informat al pacienților.
Pregătire pacient:
pacienții nu trebuie să fumeze, să mănânce sau să bea (exceptând apă), să se spele pe dinți sau să mestece gumă cu cel puțin 1 oră înaintea recoltării probei.
Specimen recoltat: tampon (swab) bucal
Recipient de recoltare: kit Adventia (contine 2 tampoane bucale)
Cauze de respingere a probei: nerespectarea condițiilor de recoltare
Stabilitate probă: probele sunt stabile 14 zile la temperatura camerei.
Metoda – NGS, MLPA, PCR, Sanger. Este evaluat un panel de 231 gene asociate bolilor genetice autozomal recesive și X-linkate pentru identificarea variantelor nucleotidice, variantelor indel mai mici de 21 perechi de baze și variații ale numărului de copii (CNV) cu dimensiunea minima de 1 exon.
ADN-ul este izolat și se efectuează secvențierea tuturor exonilor codificatori și a regiunilor intronice conservate. Sunt identificate modificările unei singure perechi de baze (SNV), micile deleții sau duplicații și variațiile numărului de copii (CNV).
Acest test permite analiza a până la 231 de boli genetice autozomal recesive și X-linkate recesive, printre care: alfa-talassemia, hemoglobinopatiile, fibroza chistică, distrofinopatiile, sindromul Fragile X, atrofia musculară spinală, anemia Fanconi tip C, boala Niemann-Pick tip A/B, deficitul de fenilalanin hidroxilază, precum și multiple alte afecțiuni cardiovasculare, hematologice, imunologice, metabolice, musculare, neurologice, pulmonare, renale și oftalmologice. Aceste patologii prezintă forme de evoluție moderată până la severă și determină dizabilitate semnificativă, afectare organică majoră, retard de dezvoltare sau reducerea speranţei de viaţă, şi de cele mai multe ori nu există tratament curativ sau managementul lor este complex şi de lungă durată.
Recomandări pentru efectuarea testului de screening Adventia - Panel genetic de bază (core) pentru identificarea statusului de purtător (carrier screening)
- Orice cuplu care planifică o sarcină, indiferent de originea etnică sau istoricul familial și care dorește informații legate de riscul genetic reproductiv
- Persoane sau cupluri aflate într-un program de reproducere asistată, inclusiv FIV
- Donatori și primitori de spermă sau ovocite
- Persoane cu istoric familial cunoscut de boală genetică recesivă (mai ales rude de gradul I sau II), după un consult genetic prealabil
- Persoane provenind din grupe de populații cu prevalență crescută a unor boli genetice
- Femei însărcinate care doresc să afle propriul status genetic și al partenerului, dacă acesta nu a fost efectuat înainte de concepție și doar împreună cu consilierea genetică. Testarea în sarcină identifică doar statusul de purtător al părinților și nu al fătului. În cazul în care ambii părinți sunt purtători ai aceleiași gene, atunci se recomandă diagnosticul prenatal al fătului. Testul de screening Adventia nu inlocuiește diagnosticul prenatal.
- Orice persoană care dorește să afle mai multe despre profilul său genetic
Adventia a fost conceput ca un test benefic și comprehensiv, destinat tuturor persoanelor, indiferent de originea etnică sau de istoricul familial. Testul se bazează pe o tehnologie inovatoare și performantă și oferă rezultate relevante într-un interval scurt de timp, contribuind la reducerea riscului de transmitere a bolilor genetice către descendenți.
Consilierea genetică înainte și după testare este esențială pentru interpretarea adecvată a rezultatelor și orientarea deciziilor reproductive, deoarece testul nu înlocuiește consultul de specialitate, iar concluziile trebuie integrate în contextul clinic al pacientului, incluzând istoricul familial, originea etnică și datele obținute din alte investigații complementare.
Adventia utilizează o tehnologie avansată de tip Target Capture Enrichment, care permite captarea selectivă a fragmentelor de ADN din regiunile de interes, asigurând acoperirea completă a exonilor pentru genele analizate și detecția simultană a variantelor SNV (variante punctiforme), INDEL (inserții/deleții) și CNV (variații ale numărului de copii) până la nivel de exon unic. Performanța metodei este susținută de o sensibilitate și specificitate foarte ridicate, confirmate prin studii de validare ale metodei și prin o analiză bioinformatică optimizată pentru a crește acuratețea. Testul Adventia prezintă o sensibilitate și specificitate clinică de 100% pentru varianta SNV/INDEL (25/25 TP (True Positive), respectiv 9945/9945 TN (True Negative)), precum și o sensibilitate de 100% și o specificitate de 99,99% pentru detecția variațiilor de număr de copii (CNV ≥2 probe), aceste rezultate evidențiind un nivel ridicat de acuratețe și diferențiind Adventia de alte paneluri comerciale disponibile.
Clasificarea variantelor identificate se realizează în conformitate cu ghidurile actuale ACMG/AMP, iar în cazul testării ambilor părinți, testul Adventia permite identificarea riscului de heterozigot compus, prin detectarea unor variante patogene diferite în cadrul aceleiași gene. Abordarea este aliniată cu recomandările majore internaționale, inclusiv ACMG 2021 pentru Expanded Carrier Screening, ACOG 2017 (reafirmat în 2023), ESHG și declarația comună ACOG/ACMG/NSGC/PQF/SMFM, susținând luarea unor decizii clinice informate, precum alegerea unui donator compatibil, utilizarea IVF cu PGT-M, realizarea diagnosticului prenatal, inițierea intervențiilor precoce pentru bolile tratabile și oferirea unei consilieri genetice personalizate.
În urma evaluării și clasificării interne, sunt raportate doar variante cu semnificație clinică patogenă, potențial patogenă sau incertă, respectiv clasele ACMG 5 (P-Pathogenic), 4 (LP-Likely Pathogenic) sau 3 (VUS-Variant of Uncertain/Unknown Significance).
Gene analizate: ABCD1, ACAD9, ACADM, ACOX1, ACSF3, ADAMTS2, AGA, AGL, AGPS, AGXT, AIRE, ALDH3A2, ALDOB, ALG6, ALMS1, ALPL, AMT, AQP2, ARSA, ASL, ASNS, ASPA, ASS1, ATM, ATP6V1B1, ATP7B, BBS1, BBS12, BCKDHB, BCS1L, BLM, BSND, BTD, CAPN3, CBS, CEP290, CERKL, CFTR, CHM, CHRNE, CIITA, CLN3, CLN5, CLN6, CLN8, CLRN1, CNGB3, COL4A3, COL4A5, COL7A1, CPT1A, CPT2, CRB1, CTNS, CTSK, CYBB, CYP11B2, CYP19A1, CYP27A1, DCLRE1C, DHCR7, DHDDS, DLD, DMD, DNAH5, DNAI1, DNAI2, DYSF, EDA, EIF2B5, ELP1, EMD, ESCO2, ETFA, ETHE1, EYS, F11, F5, F9, FAH, FAM161A, FANCC, FANCG, FMR1, G6PC, GAA, GALC, GALK1, GALT, GBA, GBE1, GCDH, GFM1, GJB2, GJB6, GLA, GLDC, GNE, GNPTAB, GNPTG, GNS, GRHPR, HADHA, HAX1, HBA1, HBA2, HBB, HEXA, HEXB, HJV, HGSNAT, HLCS, HMGCL, HOGA1, HPS1, HPS3, HSD17B4, HYAL1, HYLS1, IDS, IL2RG, IVD, LAMC2, LCA5, LDLR, LHCGR, LHX3, LIFR, LIPA, LOXHD1, LPL, LRPPRC, MCCC1, MCCC2, MCOLN1, MEFV, MFSD8, MKS1, MLC1, MMAA, MMAB, MMACHC, MMADHC, MPI, MPV17, MTM1, MTRR, MTTP, MMUT, NAGLU, NAGS, NBN, NDUFAF5, NDUFS6, NPC1, NPC2, NPHS1, NPHS2, NR2E3, NTRK1, OAT, OPA3, OTC, PAH, PCDH15, PDHA1, PDHB, PEX1, PEX10, PEX2, PEX6, PEX7, PFKM, PHGDH, PKHD1, PMM2, PPT1, PROP1, PSAP, PTS, PUS1, PYGM, RAB23, RAG2, RAPSN, RARS2, RLBP1, RMRP, RPGR, RS1, SACS, SAMHD1, SEPSECS, SGCA, SGCB, SGCG, SLC12A6, SLC17A5, SLC25A13, SLC25A15, SLC26A2, SLC26A4, SLC35A3, SLC37A4, SLC4A11, SLC6A8, SLC7A7, SMARCAL1, SMN1, SMN2, SMPD1, STAR, SUMF1, TFR2, TGM1, TH, TMEM216, TPP1, TRMU, TSFM, TTPA, TYMP, UGT1A1, USH1C, USH2A, VPS13A, VPS45, VPS53, VRK1, VSX2, WNT10A.
Interpretarea rezultatelor
1. Rezultat pozitiv: Sunt raportate și descrise variantele identificate din clasele de patogenitate 5 sau 4 care sunt asociate bolilor cu transmitere autozomal recesiv și/sau X-linkate.
2. Rezultat negativ: Nu a fost identificata nicio variantă raportabilă. În acest caz este improbabil ca tabloul clinic al pacientului sa fie asociat variațiilor genelor analizate.
3. Rezultat neacționabil: A fost identificata o varianta de tip VUS, aceasta însemnând că dovezile din literatura actuală de specialitate sunt insuficiente sau contradictorii, pentru a încadra varianta într-o clasa de patogenitate sau benignitate. Acest tip de variante sunt neacționabile și neinformative, dar sunt raportate în vederea reevaluării periodice a semnificației clinice, fiind probabilă / posibilă o reclasificare la 12 luni, în baza noilor contribuții la literatura de specialitate.
Sunt analizate și clasificate toate variantele identificate, inclusiv cele care nu sunt prezente în bazele de date specializate. Variante rare, variante de novo sau variante nepublicate în bazele de date utilizate nu sunt detectate.
Limitări și interferențe
Testul acoperă doar genele evaluate în cadrul analizei. Metoda aplicată nu poate evalua variantele structurale echilibrate (translocații, inversii) la nivelul genelor analizate.
Limitări generale ale tehnologiei NGS utilizate în carrier screening (nu pot fi detectate variante nucleotidice cu dimensiuni cuprinse intre 21 perechi de baze și 1 exon (~150pb), variantele de tip deleție/duplicație cu dimensiuni de cel puțin 1 exon necesită confirmare ulterioară ceea ce poate avea consecință prelungirea termenului de eliberare al rezultatului).
Testul nu identifică modificări ale numărului de cromozomi sau rearanjamente cromozomiale de mari dimensiuni și nici afecțiuni autozomal dominante caracterizate prin penetranță redusă.
Un rezultat negativ nu poate exclude cu certitudine o variantă neidentificabilă prin metoda de testare utilizată.
Variantele cu specificație incertă (VUS) pot fi raportate, dar clasificarea lor poate fi actualizată în funcție de publicațiile apărute în literatura de specialitate.
Bolile genetice autozomal recesive și X-linkate.
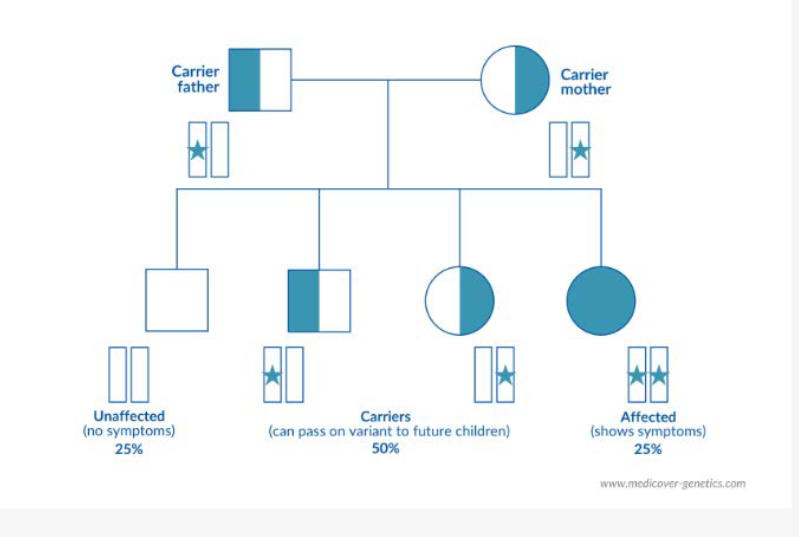
Bolile genetice sunt afecțiuni cauzate de modificări ale materialului genetic, fie la nivelul unei singure gene (mutații genice), fie la nivelul structurii sau numărului cromozomilor (aberații cromozomiale). Bolile autozomal recesive și X-linkate reprezintă două subtipuri importante ale bolilor monogenice. Primele sunt cauzate de mutații ale unor gene situate pe autozomi (cromozomi non-sexuali, din cele 22 de perechi), iar cele din a doua categorie sunt determinate de mutații ale genelor localizate pe cromozomul X. Aproximativ 1 din 4 indivizi din populația generală este purtător asimptomatic al unei variante patogene asociate unei afecțiuni autozomal recesive.
În cazul bolilor autozomal recesive, indivizii pot fi fie purtători sănătoși ai mutației (heterozigoți) — cu o singură copie mutantă a genei și fără manifestări clinice — fie persoane la care boala se exprimă complet, fie prin prezența a două copii identice ale variantei patogene (homozigot), fie prin prezența a două variante patogene diferite la nivelul aceleiași gene (heterozigot compus).
Prezența unei singure variante patogene nu este suficientă pentru manifestarea clinică a bolii. Copia funcțională rămasă a genei compensează efectul variantei patogene, astfel încât purtătorii heterozigoți sunt asimptomatici. Datorită acestui mecanism, variantele patogene asociate bolilor recesive pot fi transmise de-a lungul mai multor generații fără nicio manifestare clinică.
Riscul de a avea un copil afectat apare exclusiv atunci când ambii părinți sunt purtători ai unei variante patogene în aceeași genă, iar copilul moștenește câte o copie mutantă de la fiecare dintre ei. Bolile autozomal recesive afectează în mod egal bărbații și femeile, deoarece genele implicate sunt situate pe autozomi, nu pe cromozomii sexuali.
Dacă ambii părinți sunt purtători există risc de ¼ (25%) să aiba un copil afectat (homozigot sau heterozigot compus), ½ (50%) risc să aibă un copil purtător (asimptomatic, cu risc de transmitere la generația următoare) si ¼ (25%) să aibă un copil sănătos, nepurtător.
Boala genetică autozomal recesivă
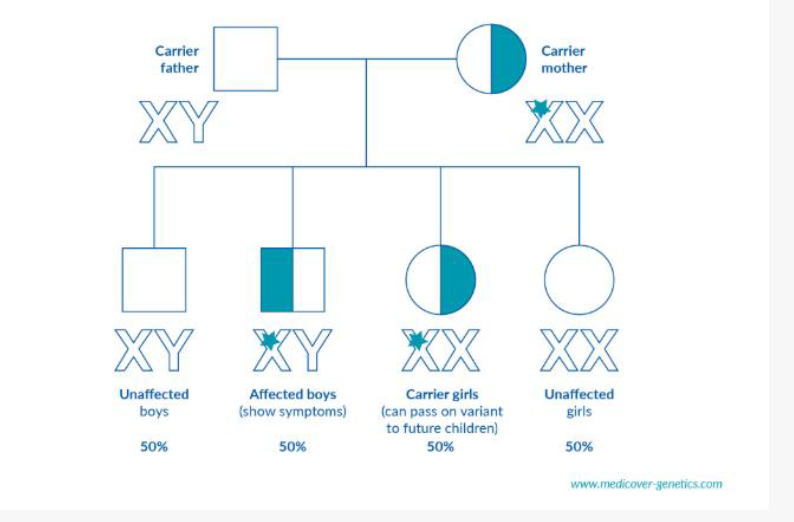
În cazul bolilor genetice X-linkate, datorită localizării genei mutante pe cromozomul X, transmiterea şi expresia clinică diferă semnificativ în funcţie de sex. Deoarece bărbații au un singur cromozom X (genotip XY), prezența unei singure alele (copii) mutante este suficientă pentru apariția bolii, astfel bărbații care moștenesc gena vor avea întotdeauna boala manifestată clinic.
Femeile (XX) trebuie să moștenească două copii mutante pentru a fi afectate; altfel, ele devin de obicei purtătoare sănătoase, fără simptome sau cu manifestări foarte ușoare datorită existenței unei copii normale a genei, în functie de care cromozom X este activ. În cel de-al doilea caz vorbim de purtătoare manifeste (manifesting carriers – fenotip parțial sau complet) la care unul dintre cei doi cromozomi X este inactivat aleatoriu, iar cromozomul X predominant activ este purtătorul de variantă patogenă (silențiere genică).
Mamele purtătoare au un risc de 50% ca fiii lor să fie afectați și un risc de 50% ca fiicele lor să devină purtătoare. Tații afectați transmit cromozomul X modificat tuturor fiicelor, care devin purtătoare, dar niciunui fiu, deoarece fiii primesc cromozomul Y.
Boala genetică legată de cromozomul X
Deoarece purtătorii sunt asimptomatici, aceștia nu sunt conștienți de statusul lor genetic și de riscul de a transmite mutația copiilor. În absența testării genetice, nu este posibilă determinarea statusului de purtător al unei boli recesive.
Bibliografie
- Medicover Genetics. Adventia Carrier Screening Panel – Ghid clinic, versiunea 1.0.
- Medicover Genetics. Carrier Screening. [Internet]. Disponibil la: https://medicover-genetics.com/our-genetic-tests/reproductive-health/carrier-screening/
- Lazarin GA, Haque IS, Evans EA, Goldberg JD. An empirical estimate of carrier frequencies for 400+ causal Mendelian variants: results from an ethnically diverse clinical sample of 23,453 individuals. Genet Med. 2013;15(3):178–186.
- Lee K, Abul-Husn N, Amendola L, et al. ACMG SF v3.3 list for reporting of secondary findings in clinical exome and genome sequencing: a policy statement of the American College of Medical Genetics and Genomics (ACMG). Genetics in Medicine. 2025. PMID: 40568962
- ACMG updates guidance on carrier screening. Am J Med Genet A. 2022;188:9-10. doi:10.1002/ajmg.a.62267
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-24. PMID:25741868.
- Tavtigian SV, Harrison SM, Boucher KM, Biesecker LG. Fitting a naturally scaled point system to the ACMG/AMP variant classification guidelines. Hum Mutat. 2020;41:1734-7. doi:10.1002/humu.24088
- Pejaver V, Byrne AB, Feng BJ, Pagel KA, Mooney SD, Karchin R, O’Donnell-Luria A, Harrison SM, Tavtigian SV, Greenblatt MS, Biesecker LG, Radivojac P, Brenner SE; ClinGen Sequence Variant Interpretation Working Group. Evidence-based calibration of computational tools for missense variant pathogenicity classification and ClinGen recommendations for clinical use of PP3/BP4 criteria. Am J Hum Genet. 2022;109(12):2232-2247. doi:10.1016/j.ajhg.2022.10.013.
- Miller DT, Lee K, Gordon AS, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2021 update: a policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2021;23(8):1391-8.
- Gordon AS, Lee K, Abul-Husn NS, et al. Consideration of disease penetrance in the selection of secondary findings gene-disease pairs: a policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2024;26(7):101142.
- Kalia SS, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2017;19(2):249-55.
- ACMG Board of Directors. The use of ACMG secondary findings recommendations for general population screening: a policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2019;21:1467-8. doi:10.1038/s41436-019-0502-5.
- O’Daniel JM, et al. A survey of current practices for genomic sequencing test interpretation and reporting processes in US laboratories. Genet Med. 2017;19:575-82. doi:10.1038/gim.2016.152. PMID:27811861.
Vezi tot conținutul
Vezi mai puțin